
The method of 100-kernel weight related genes mining in maize mixed with genetic algorithm and XGboost
-
摘要: 基于RNA-Seq的转录组测序数据特征维度较高,使用传统生信方法寻找表型相关基因需要大量计算资源,且差异分析所得候选基因范围较大,进一步筛选依赖已有的先验知识。针对这一问题,本文提出了融合遗传算法和XGBoost的转录组分析方法—GA-XGBoost,通过融入机器学习算法缩小了后续分析的候选基因范围。在一组高质量玉米数据集上对基因–百粒重性状的关联进行了对比实验和后续分析,结果显示,相比于分别使用全体基因和差异表达基因直接训练XGBoost模型,所提方法得到的候选基因训练的XGBoost模型在玉米百粒重的预测结果上具有最小的MSE;相比于差异表达分析结果的1542个差异表达基因,GA-XGBoost方法最终将候选基因范围减小至48个,范围缩小了31倍,表明所提方法能够有效提升对转录组数据的分析能力和效率。Abstract: The RNA-Seq-based transcriptome sequencing data has a high feature dimension that requires a lot of computing resources when using traditional methods to find phenotype related genes. Moreover, the range of candidate genes obtained by difference analysis is large, and further screening depends on existing a prior knowledge. A transcriptome analysis method combining genetic algorithm and XGBoost, GA-XGBoost, was proposed to narrow the range of candidate genes for subsequent analysis by incorporating machine learning algorithm. A comparative experiment and subsequent analysis of the gene-100-kernel weight trait association on a set of high-quality maize datasets showed that, compared with training the XGBoost model directly with whole genes and differentially expressed genes, the candidate gene training XGBoost model obtained by the proposed method had the minimum MSE in predicting the 100-kernel weight of maize. Compared with 1542 differentially expressed genes in the results of differential expression analysis, the range of candidate genes was reduced to 48 by the GA-XGBoost method, which was reduced by 31 times, indicating that the proposed method could effectively improve the ability and efficiency of transcriptome data analysis.
-
转录组分析是一种快速有效的基因组调查、大规模功能基因和分子标记鉴定的方法[1]。相较于基因芯片等方法,基于转录组测序(RNA-sequencing, RNA-Seq)的方法不依赖基因的先验知识,能够覆盖更大的转录组范围,具有更高的分辨率并且测序成本更低[2]。已有很多学者针对RNA-Seq测序数据进行了研究[3- 4],其中不乏使用机器学习进行研究的方法[5- 6]。通过RNA-Seq得到的转录组测序数据具有样本量较少(几十或者几百个)、基因数极高(通常有上万个基因)的特点。数据高维的特点导致对其进行分析需要更大的计算资源和时间;同时,传统的统计方法往往也由于数据维度过高而失效。因此,对数据进行降维,寻找能够表示其特征空间的最优子集成为研究人员需要解决的问题。
常见的转录组分析方法主要可以分为两类:1) 根据已知的生物学领域知识和统计知识对数据进行处理,筛选出相对低维的特征空间进行后续研究,例如差异表达分析。此类方法[7- 8]能够较快速地获得特征子空间,但是无法保证子空间能够保留原始空间的全部信息,从而可能导致最终的效果不尽如意。2) 结合机器学习算法,从样本的基因全集中选择若干个基因作为特征构建学习器,并根据学习器的性能和基因在学习器中的重要性(如特征权重)筛选候选基因[5]。此类方法使用学习器的性能作为评判标准,虽然能够获得比较优秀的特征子集,但是只是针对单一特征进行评价,没有考虑到基因之间的相互作用。而基因间的相互作用也会导致表型的差异,如此选出的特征子集往往不是最优子集。
遗传算法(genetic algorithm,GA)是一个在全局层面对问题寻找最优解的算法,借鉴了自然界中的物种进化规律,最早由 Holland[9]于1975 年在其专著《自然界和人工系统的适应性》中发表。遗传算法每次迭代保留一组候选解,通过模仿生物繁殖的过程产生新的候选解集。使用遗传算法搜索最优特征子空间的优势在于它不需要事先考虑相关的领域知识,并且由于每次迭代都是针对一个种群进行整体评价,因此能够考虑特征间的相互作用。目前,在各个领域均有学者利用遗传算法进行研究并发表了文章。例如,文献[10]提出了一种结合多目标优化和精英策略的遗传算法,对改进的van-Genuchten (VG)模型进行升级,提高了生物炭改良土壤保水模型的预测能力。文献[11]中基于遗传算法设计的自动化方法,能够优化建筑调查激光扫描仪定位网络,最小化点云数据间的重叠。文献[12]将遗传算法用于面部识别领域,并在微表情识别上取得较好的效果。此外,在能源[13]、医疗[14]、生物信息学[15]等方面,也能发现相关研究,表明遗传算法是一个相当成熟的方法,可以应用于多个方面。然而,遗传算法对适应性度量的定义具有很大的依赖性,不同的适应度定义标准可能会导致最终得到的结果差异巨大。
本文着眼于玉米的转录组测序数据,以挖掘影响玉米百粒重性状的候选基因作为切入点进行研究。挖掘候选基因的过程本质上是特征选择的过程,即从包含全部基因的特征全集中提取部分基因组成一个特征子集,同时保证使用该子集构建的模型相比于使用全集构建的模型具有更出色的性能。通过融合遗传算法与XGBoost,对高质量玉米转录组测序数据及其产量数据进行分析,得到了调控玉米百粒重性状的候选基因。在模型的准确性方面,将所用模型与分别采用全体基因和差异表达基因(differentially expressed genes,DEGs)进行训练的XGBoost模型进行了比较。
此外,传统生信分析往往在获得差异基因后直接进行高层分析,这意味着需要从大量候选基因中进行筛选,任务量繁重而且依赖生物学先验知识。本文方法通过机器学习算法筛选差异基因,有效缩小了候选基因范围,并且不依赖先验知识。
1. 基因定量和差异表达分析
本文的整体分析方法流程如图1所示,首先对RNA-Seq数据进行基因定量,根据基因表达量进行差异表达分析。之后,使用GA-XGBoost根据差异基因和表型数据选择基因子集,通过对XGBoost模型中特征重要性排序获得候选基因。最后,对所得候选基因进行功能注释以验证方法有效性。
 图 1 方法整体流程Fig. 1 Method overall process
图 1 方法整体流程Fig. 1 Method overall process 下载:
全尺寸图片
下载:
全尺寸图片
1.1 基因定量
刚脱机的fastq格式转录组测序原始数据中主要包含若干条碱基读段(read)和其对应的标识信息,开展研究之前,首先需要确定每个读段由哪一个基因转录而来,该基因翻译了几次,这个过程称为基因定量。基因定量主要包括3个步骤。1) 质控,对测序数据进行筛选,去除测序样本中长度异常的碱基读段以及样本中的衔接子(adaptor)序列,提高基因定量的准确性。2) 构建索引,根据参考基因组及其注释文件获得测序物种的外显子和剪切位点信息,构建索引文件,作为碱基序列比对的模板。3) 定量,通过适当的比对算法,逐一将测序数据中的读段与全部基因比对,确定读段的来源,每次确定读段来源时,其对应的基因表达计数加一。完成基因定量后,每一个样本都会得到一个基因表达矩阵,其中记录了不同基因的表达信息。
一个样本组织中,一定量的RNA中转录本的量是固定的,但使用高通量测序技术对样本进行建库测序时并不能确定一共有多少转录本,对所测数据进行比对分析所得的基因的表达水平只是相对的定量;此外,基因长度也会影响转录本的读段数量,基因越长,其对应的表达次数往往也就越高。另一方面,比较基因在样本间的表达水平时,由于不同样本往往对应不同的测序深度,测序深度更深的样本会得到更多的读段,导致其基因的表达计数更高。因此,在比较不同样本间的基因表达水平之前,需要寻找一种对数据进行标准化处理的方法,消除基因表达定量过程中由于基因长度与测序深度不同而产生的差异。常用的基因表达量标准化方法有计算其每百万碱基读段数[16-17](reads per kilobase per million, RPKM)值或每百万碱基片段数[18-19](fragments per kilobase per million, FPKM)值:
$$ {\rm{RPKM}}=\frac{{10}^{6}\times {n}_{r}}{L\times N} $$ (1) $$ {\rm{FPKM}}=\frac{{10}^{6}\times {n}_{f}}{L\times N} $$ (2) 式中:
$ {n}_{r} $ 、$ {n}_{f} $ 分别为比对至目标基因的读段(read)、片段(fragment)数量;$ L $ 为目标基因的外显子长度之和除以1000, 单位是千碱基(Kb);$ N $ 为比对至基因组的有效读段总量。二者的区别在于,RPKM是采用读段的数量进行标准化,FPKM采用的则是片段的数量。当测序方式为双端测序(pair-end,PE)时,一个片段对应两个读段;但当测序方式为单端测序时,使用RPKM与FPKM进行标准化的结果没有区别。1.2 差异表达分析
一个生物学个体或者组织,在不同的生长发育周期、不同的组织细胞中,其存在的全体基因并非全部表达,而是根据实际需要部分表达。因此,不同组织或统一组织在不同发育周期中基因的表达模式存在差异,有的基因大量表达行使功能,有的基因少量表达,另一部分基因则完全不表达。往往,具有相同性状的个体或组织间,其基因的表达模式相同,性状相差较大的个体之间,基因的表达模式差别也较大。基于该情况,在通过测序得到基因的表达信息之后,可以根据样本性状的分组信息,通过统计学方法对其表达模式进行分析,寻找样本间差异表达的基因,进而缩小影响性状的基因范围。
差异表达分析的缺陷在于,其分析方法依赖样本的分组信息;另一方面,生物学者进行实验设计时,往往只针对目标性状进行统计,但检测到的差异基因同时包括其他无关性状的相关基因,这也是导致最终得到的差异表达基因较多的原因,需要后续针对目标性状进行深入分析以筛选候选基因。
2. 基于GA-XGBoost算法的特征选择
2.1 XGBoost算法
XGBoost(eXtreme gradient boosting)是陈天奇[20]于 SIGKDD 2016 大会上提出的一种基于梯度提升和决策树的集成学习方法,能够有效地学习数据间的关系。
XGBoost使用分类和回归树(classification and regression trees,CART)作为基学习器,通过在损失函数中引入正则项控制模型的复杂度以确保泛化能力,正则项包括叶子节点数和叶子节点权重的平方和。XGBoost的思想是每次增加一棵树拟合上一轮预测结果的残差,通过不断地增加新树达到降低损失值的目的,基于加法模型将多个弱学习器集成为一个强学习器,从而获得一个具有高准确率的机器学习模型。
对于一个包含
$ n $ 个$ m $ 维特征样本的数据集$D= $ $ \left\{\left({\mathit{x}}_{1},{y}_{1}\right),\left({\mathit{x}}_{2},{y}_{2}\right),\cdots ,\left({\mathit{x}}_{\mathit{n}},{y}_{n}\right)\right\}$ ,对于第$ i $ 个输入$ {\mathit{x}}_{\mathit{i}} $ ,XGBoost预测的输出为$$ {\widehat{y}}_{i}=\text{Ø} \left({\mathit{x}}_{\mathit{i}}\right)=\sum _{k=1}^{K}{f}_{k}\left({\mathit{x}}_{\mathit{i}}\right) $$ (3) $$ {f}_{k}\in \mathcal{F},\mathcal{F}=f\left(x\right)={\omega }_{q\left(x\right)} $$ (4) 式中:
$ {f}_{k} $ 表示第$ k $ 个分类和回归树,取值范围为$ [1,K] $ ;$ {\omega }_{q\left(x\right)} $ 为分类和回归树对样本结果的预测值;$ K $ 为模型中设置的分类和回归树的总数。XGBoost需要优化的目标函数为
$$ L\left(\text{Ø}\right)=\sum _{i}l\left({\widehat{y}}_{i},{y}_{i}\right)+\sum _{k}\varOmega \left({f}_{k}\right) $$ (5) $$ \varOmega \left({f}_{k}\right)=\mathrm{\gamma }T+\frac{1}{2}\lambda \sum _{j=1}^{T}{\omega }_{j}^{2} $$ (6) 式中:
$ l\left({\widehat{y}}_{i},{y}_{i}\right) $ 为预测值关于真实值的损失函数;$\varOmega \left({f}_{k}\right)$ 为目标函数的正则项;$ \mathrm{\gamma } $ 、$ \lambda $ 为惩罚项的系数;$ T $ 为第$ k $ 棵树的叶子结点总数;$ {\omega }_{j} $ 为树上第$ j $ 个叶子节点的输出分数,$ j $ 的取值范围为$ [1,T] $ 。在XGBoost的应用上,文献[21]将XGBoost与深度学习结合,建立了对肝细胞癌微血管侵犯(microvascular invasion,MVI)的术前鉴定模型;文献[22]使用XGBoost在肺癌的检测和复发预测方面进行了系统研究。文献[23]从基因表达数据入手,对20种实体瘤的原发灶进行了推断;文献[24]使用XGBoost辅助识别RNA上的N6-甲基腺苷(N6-methyladenosine)位点。这些研究表明XGBoost适用于本文的研究内容。
然而,一般情况下XGBoost模型的效果受到所用训练数据规模的影响,当训练数据集中每一条数据的特征维度都很高但样本总数又相对较少时,如转录组数据,XGBoost往往难以学习到数据中的全部信息,模型的预测效果就会较差。如果能够在训练之前,通过特征工程等实现特征筛选,然后训练模型,便能够显著提高XGBoost模型的预测效果,这将在后面的表2中有所体现。因此,如何对输入数据进行预处理从而提高XGBoost模型的效果是一个需要考虑的问题。
2.2 GA-XGBoost算法
在生物体中,不同的基因负责调控不同的性状,因此,挖掘玉米百粒重性状候选基因的过程本质上是特征选择的过程。将样本包含的全部基因视为特征全集,从中提取部分基因构成一个特征子集,同时保证使用该子集构建的模型相比于使用全集构建的模型具有更出色的性能。
遗传算法是模拟生物界种群演化规律的随机搜索算法,主要借鉴种群繁殖过程中个体杂交、染色体交换和基因突变的情况,根据一定的规则模仿自然选择生成新一代种群,并通过不断的重复该过程从而找到最适应环境的最优种群。在遗传算法中,问题可能的一个解叫做个体,通过一定的编码规则转换为一个唯一的向量表示,称作染色体,一组可能的解构成一个种群。使用遗传算法进行特征选择时,个体适应度的设定对最终所得最优子集具有重要的影响,不够合理的适应度设定将导致最终的子集并非最优解。本文将遗传算法与XGBoost融合,提出遗传算法–极限梯度提升算法(genetic algorithm-XGBoost,GA-XGBoost),解决了遗传算法中个体适应性度量的设定问题和XGBoost需要对输入数据进行预处理的问题,并且保留了两个算法各自的优点。
基于遗传算法-XGBoost(GA-XGBoost)的特征选择方法包含个体编码、种群初始化、自然选择、染色体交叉和基因突变、迭代结束判断等步骤。GA-XGBoost算法的流程如图2所示。
 图 2 GA-XGBoost算法Fig. 2 GA-XGBoost algorithm下载:
全尺寸图片
图 2 GA-XGBoost算法Fig. 2 GA-XGBoost algorithm下载:
全尺寸图片
G表示算法设定的迭代次数,伪代码部分见算法1。
算法 1 GA-XGBoost
输入 全特征数据集;
输出 最优特征子集。
1) 初始化种群矩阵和迭代次数G;
2) for g = 0; g < G; G = g + 1 do:
3) 利用XGBoost计算个体适应度;
4) 根据适应度随机选择个体;
5) 染色体交叉;
6) 随机基因突变;
7) 生成新种群;
8) if 连续n代个体平均适应度不变;
9) break
10) end for
11) return 优化特征子集。
1) 个体编码
遗传算法中,个体指所求问题对应的一组可能的解,编码指通过一定的规则将个体转换为唯一的向量表示。编码后的向量称作染色体,向量中的每一位称作一个基因。
对于
$ n $ 维特征选择的问题,每一个候选特征对应一个基因,个体表示为一个形如$\left({x}_{1},{x}_{2},\cdots ,{x}_{n}\right)$ 的$ n $ 维向量,其中$ {x}_{i}\in \left\{0,\mathrm{ }1\right\} $ ,$ {x}_{i}=1 $ 表示选中第i个特征,否则$ {x}_{i}=0 $ 。如图3所示,对于含有6个特征的集合$ \left\{{f}_{1},{f}_{2},{f}_{3},{f}_{4},{f}_{5},{f}_{6}\right\} $ ,个体$ \left(\mathrm{1,0},\mathrm{0,0},\mathrm{1,1}\right) $ 表示所选特征子集为$ \left\{{f}_{1},{f}_{5},{f}_{6}\right\} $ 。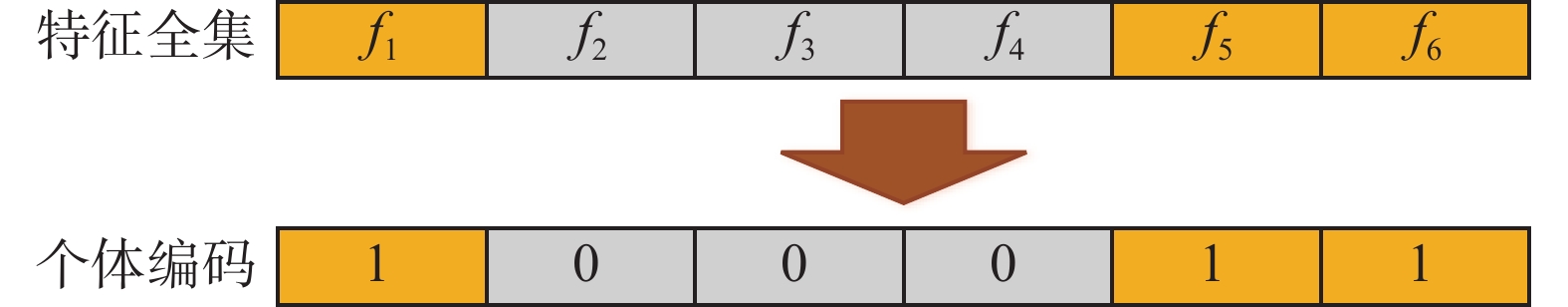 图 3 特征编码Fig. 3 Feature coding下载:
全尺寸图片
图 3 特征编码Fig. 3 Feature coding下载:
全尺寸图片
2)种群初始化
遗传算法中种群指多组候选解的集合。此处将种群大小设置为50,个体每个基因位编码从
$ \left\{0,\mathrm{ }1\right\} $ 中按照等概率随机抽取。3) 自然选择
自然选择的过程需要考虑个体对环境适应性度量的问题。在特征选择的问题上,遗传算法中个体的适应度表现为该个体染色体上的基因信息(即选择的特征)对预测值的影响。
具体地讲,使用XGBoost建立回归树模型,对种群中每个个体的染色体分别解码,根据个体所选的特征对实验数据训练集进行训练,并在测试集上根据模型的均方误差(mean square error,MSE)度量个体的适应性。均方误差的计算公式为
$$ {\rm{MSE}}=\frac{1}{M}{\sum_{i=1}^{M}}{\left({y}_{i}-\widehat{{y}_{i}}\right)}^{2} $$ (7) 式中:
$ M $ 为测试集样本的总数;$ {y}_{i}、\widehat{{y}_{i}} $ 分别为第$ i $ 个样本的真实值和模型的预测值。由式(7)可知,MSE越小表明模型的性能越高。然而,适应度要求个体对环境适应性越好,则适应度越高。因此,为了保证对环境适应性更好的个体具有更高的适应度,第$ i $ 个个体的适应度最终定义为$$ {\rm{fitness}}\left(i\right)=100-{\rm MSE}_{i} $$ (8) ${\rm MSE}_{i}$ 表示使用第$ i $ 个个体中所选特征训练的XGBoost在训练集上的均方误差。值得注意的是,XGBoost的参数设置在每轮种群间迭代以及个体计算适应度时保持不变,从而保证算法不会由于XGBoost模型的参数问题导致对不同个体的适应度计算产生差异。根据自然选择的规律,种群中适应度大的个体理应有更大的概率保留下来并繁殖下一代,即个体被选中的概率与其适应度成正相关。对于大小为
$ s $ 的种群$ P $ ,设其中第$ i $ 个个体$ {idv}_{i} $ 的适应度为${\rm{fitness}}\left(i\right)$ 。为了确保算法的稳定性,首先选择种群中适应度排名最高的$ k $ 个个体,对于剩下的$(s-k)$ 个位置,根据式(2)~(7)计算保留的概率并以此为依据从全部个体中进行随机选择。$$ p\left(i\right)=\dfrac{{\rm{fitness}}\left(i\right)}{{\displaystyle\sum_{k=1}^{s}}{\rm{fitness}}\left(k\right)} $$ (9) 4) 交叉和突变
遗传算法中的交叉和突变步骤,模拟的是生物界个体繁殖产生子代的过程。具体来说,针对
$ n $ 维特征选择的问题,长度为$ {l}_{f} $ 的部分父亲染色体和长度为$ {l}_{m} $ 的部分母亲染色体结合,产生子代染色体,其中$ {l}_{f}+{l}_{m}=n $ ,该过程称作交叉,两部分染色体结合的位置随机产生,称作交配点。同时,繁殖过程中还可能发生基因突变的情况,即染色体中某一个基因的值发生反转,从0至1或者从1至0。图4为本文算法中交叉和突变的示例图。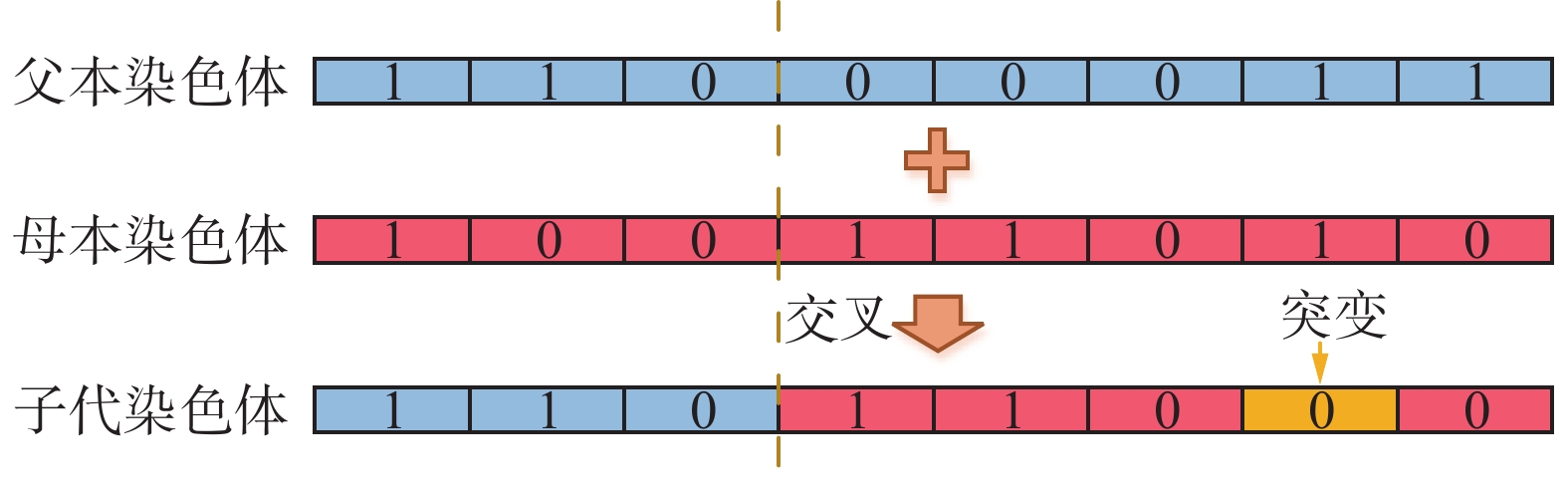 图 4 交叉和突变Fig. 4 Crossover and mutation下载:
全尺寸图片
图 4 交叉和突变Fig. 4 Crossover and mutation下载:
全尺寸图片
需要指出的是,交叉和突变并不总是发生的。交叉的目的是为了确保子代能够产生染色体不同的新个体,从而寻找所求问题新的可能解。突变是为了避免算法陷入局部最优解,但若突变几率较大,也会导致算法在全局最优解依然无法稳定。
5) 迭代结束判断
当算法迭代至设定的轮数或者满足指定的收敛条件,比如连续多代种群中个体的平均适应度不变,即可认为种群已经成熟,此时算法终止。在最终得到的种群中,选择适应度最高的个体,对其染色体进行解码从而获得所求问题的最优特征子集。
2.3 候选基因提取
GA-XGBoost算法结束后,输出最优特征子集信息。该信息是一个编码后的向量,根据2.2节介绍的编码方法对该向量进行解码,得到与表型性状相对应的最优基因子集,之后使用该子集中基因的表达量和对应样本的表型数据训练XGBoost模型,根据模型对特征的重要性排序信息,选择排列最靠前的若干基因作为候选基因。
2.4 GA-XGBoost时间复杂度分析
使用GA-XGBoost进行数据处理分为遗传算法寻找特征和XGBoost评价个体适应度两部分。遗传算法寻找最优特征时的时间复杂度为
$ O\left(mG\right) $ ,其中$ m $ 为种群大小,$ G $ 为迭代次数,最大时间复杂度为$ O\left({G}^{2}\right) $ ;使用XGBoost计算适应度部分的时间复杂度为$O(mn{\rm log}_{2}^{n}+Kmnd)$ ,$ m $ 为特征总数,$ n $ 为样本总数,$ K $ 为XGBoost中树的总数,$ d $ 为树深度。因此,GA-XGBoost算法的时间复杂度为$O\left({G}^{2}\left(mn{\rm log}_{2}^{n}+ $ $ Kmnd\right)\right)$ 。此外,其他常用于特征选择的方法,如基于递归特征消除的支持向量机(support vector machine recursive feature elimination,SVM-RFE)[25]算法,SVM模型训练阶段的时间复杂度介于
$O\left({N}_{S}^{3}+{N}_{S}^{2}n+\right. $ $ \left.{N}_{s}mn\right)$ 和$ O\left(m{n}^{2}\right) $ 之间,其中,$ {N}_{s} $ 为模型中支持向量的个数,$ m $ 为输入向量的维度,$ n $ 为训练样本的个数;递归特征消除(recursive feature elimination, RFE)阶段需要迭代的次数为特征数$ m $ ,每次特征排序的时间复杂度为$ O\left({m}^{2}\right) $ ,因此,SVM-RFE的最大时间复杂度为$ O\left({m}^{4}{n}^{2}\right) $ 。当提取候选基因时,输入基因数据的维度往往上万,GA-XGBoost算法的时间复杂度优于SVM-RFE,并且GA-XGBoost在全局层面的特征选择能够考虑基因间的作用关系,这也是SVM-RFE无法实现的。3. 实验结果与分析
数据集采用农科院高质量的玉米转录组测序数据和对应的百粒重测产数据。对测序数据进行质控后,比对玉米B73 RefGen_v4参考基因组统计样本中基因的表达水平,并计算FPKM值,对同一实验条件下的重复样品,FPKM取所有重复数据的平均值。
3.1 差异表达分析
所用RNA-Seq数据集,经过与参考基因组比对后,共检测到539 31个基因,其中表达量不为0的基因个数有419 24个。根据样本的分组信息,使用基于R包的Deseq2对表达量非零的基因进行差异表达分析。在校验后P值(P-adjusted,padj)小于0.1的情况下,当
$ \left|\mathrm{L}\mathrm{F}\mathrm{C}\right| > 0 $ 时,共检测到934个基因表达上调,占非0基因总数的2.2%,860个基因表达下调,占非0基因总数的2.1%,共计1794个基因在两组样本间差异表达;当$ \left|\mathrm{L}\mathrm{F}\mathrm{C}\right| > 1 $ 时,共检测到843个基因表达上调,占非0基因总数的2.0%,699个基因表达下调,占非0基因总数的1.7%,共计154 2个基因在两组样本间差异表达。差异表达基因的具体信息见表1, MA图(M-versus-A plot, MA plot)见图5。3.2 基于GA-XGBoost的表型相关基因挖掘
3组对照实验中,第1组使用样本的FPKM矩阵作为输入矩阵,第2组使用只含差异基因的FPKM矩阵,同时两组均使用百粒重产量矩阵作为输出矩阵,并分别使用网格搜索将XGBoost模型调整到最优。第3组实验首先固定一组XGBoost模型的超参数,然后将该模型结合遗传算法对包含全部基因FPKM值的矩阵进行特征选择。在获得最优特征子集之后,以该特征子集作为输入矩阵训练XGBoost模型并通过网格搜索将模型调整至最优。图6所示为种群中个体适应度均值关于遗传算法迭代次数的关系,其中横轴表示遗传算法迭代次数,纵轴表示每代种群中个体适应度的均值。由图6可知算法在第40轮迭代之后开始收敛。
表 1 差异表达分析结果 (padj < 0.1,419 24个非0基因)Table 1 Differential expression analysis results (padj < 0.1,419 24 non-zero genes)差异表达
基因类型Log2FoldChange
(LFC)差异表达
基因数量差异表达
基因占比/%上调基因 LFC > 0 934 2.2 上调基因 LFC > 1 843 2.0 下调基因 LFC < 0 860 2.1 下调基因 LFC < 1 699 1.7  图 5 差异表达基因的MA图Fig. 5 MA-plot of DEGs下载:
全尺寸图片
图 5 差异表达基因的MA图Fig. 5 MA-plot of DEGs下载:
全尺寸图片
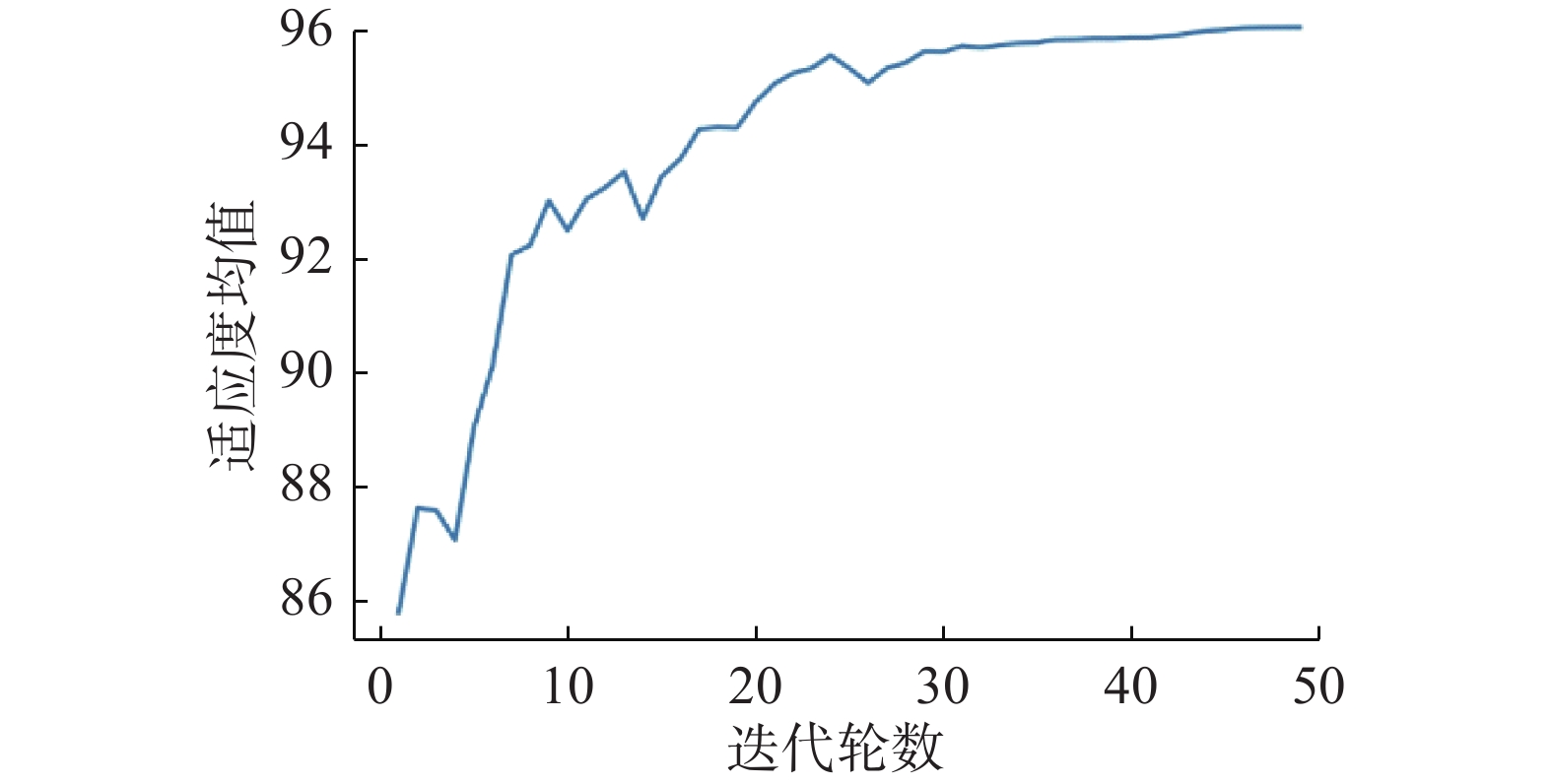 图 6 GA-XGBoost中适应度的变化Fig. 6 Change of fitness in GA-XGBoost下载:
全尺寸图片
图 6 GA-XGBoost中适应度的变化Fig. 6 Change of fitness in GA-XGBoost下载:
全尺寸图片
将3组实验得到的模型对同一个测试集进行预测,选择MSE作为比较3个模型的评判标准,最终得到的结果如表2所示。从表2易知,遗传算法-XGBoost所得回归模型在所用数据集上具有最小的均方误差,说明该模型能够最好地拟合所用数据。
表 2 3种方法的MSETable 2 MSE of three methods方法 MSE 全体基因训练所得XGBoost模型 9.183 差异表达基因所得XGBoost模型 7.689 GA-XGBoost最优子集所得XGBoost模型 3.752 对基于遗传算法-XGBoost得到的最优个体进行解码得到最优基因子集。使用该子集训练XGBoost模型并通过网格搜索确定最优参数,之后根据模型中的特征重要性排序,获得与实验样本百粒重性状关联最大的若干个候选基因。图7(a)、(b)显示了不同重要性判断指标下部分XGBoost模型中重要性较高的基因,横坐标为模型中该特征的重要性分数,纵坐标为特征名称,即基因ID。所选特征中具有代表性的有Zm00001d040166(Entrez Gene ID:103651852)和Zm00001d041800(Entrez Gene ID:103650619)等。
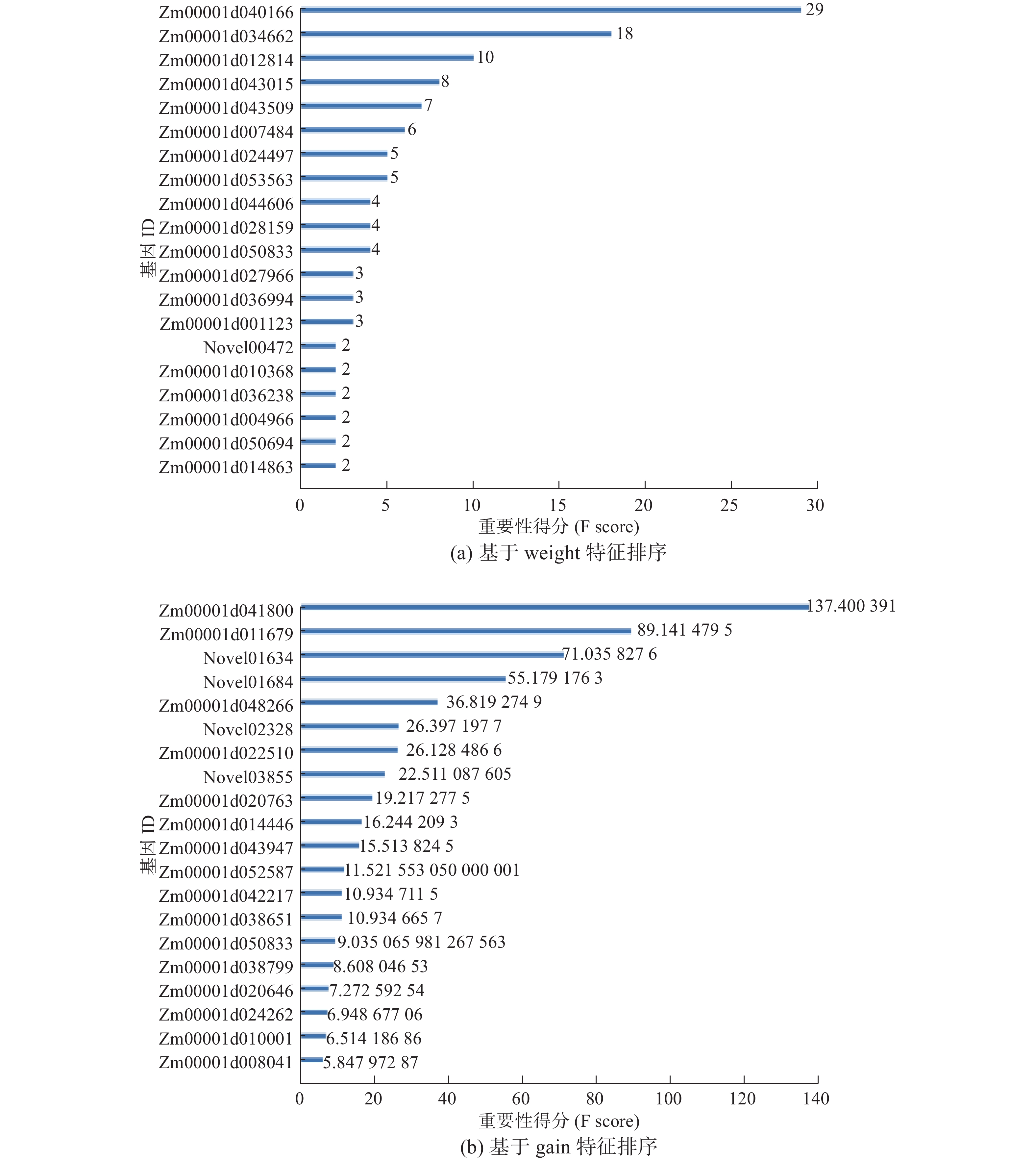 图 7 XGBoost模型中部分重要特征Fig. 7 Part of important features in XGBoost model下载:
全尺寸图片
图 7 XGBoost模型中部分重要特征Fig. 7 Part of important features in XGBoost model下载:
全尺寸图片
相较于传统生物信息学方法在进行差异表达分析之后,直接使用差异基因作为候选集合寻找目标基因的方式,通过GA-XGBoost算法筛选后的候选集排除了大量的无关基因,具有范围更小的优势,能够大幅提高寻找目标基因的效率。表3比较了针对玉米百粒重性状,经过GA-XGBoost筛选后的结果相较于传统方法的候选基因集大小差异,其中,差异表达基因的判定条件为padj < 0.1,|LFC| > 1。
表 3 候选基因数量比较Table 3 Comparison of the candidate genes number方法 候选基因数量 差异分析 154 2 GA-XGBoost 48 3.3 候选基因功能注释
对所得候选基因集,在NCBI(https://www.ncbi.nlm.nih.gov/)网站gene数据库中搜索基因信息,之后根据候选基因的Gene Symbol在David(https://david.ncifcrf.gov/home.jsp)网站进行基因本体(gene ontology,GO)注释和京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)通路分析。
对候选基因的基因本体注释结果显示,在生物过程方面,主要涉及细胞氧化还原稳态(GO:0045454)、跨膜转运(GO:0055085)、细胞壁修饰(GO:0042545)、果胶分解代谢过程(GO:0045490)、碳水化合物代谢过程(GO:0005975)、转录调控和DNA模板(GO:0006351、GO:0006355)、金属离子响应(GO:0010038)等过程;在细胞组分方面,主要涉及内质网膜(GO:0005789)、膜组成成分(GO:0016021)、细胞膜(GO:0005886)、细胞壁(GO:0005618)、细胞核(GO:0005634)等组分;在分子功能方面,主要涉及氧化还原酶活性(GO:0016491)、水解酶活性(GO:0016788)、电子转移活性(GO:0009055)、蛋白质二硫键还原酶活性(GO:0015035)、果胶酯酶活性(GO:0030599)、天冬氨酰酯酶活性(GO:0045330)、蛋白质酪氨酸激酶活性(GO:0004713)、三磷酸腺苷结合(GO:0005524)、葡聚糖转移酶活性(GO:0004134)、碳水化合物结合(GO:0030246)、蛋白丝氨酸/苏氨酸激酶活性(GO:0004674)、DNA结合(GO:0003677)、ATP酶(GO:0016887)等功能。具体的基因本体注释信息如表4~6所示。
表 4 候选基因GO注释–生物过程Table 4 GO of the candidate genes-biological process基因ID 基因说明 基因名称 生物过程 GO ID GO 注释 Zm00001d044606 Grx_S16-Glutaredoxin Subgroup Ii LOC100193989 0045454 Cell Redox Homeostasis Zm00001d011679 High Affinity Nitrate Transporter 2.5 LOC103636218 0055085 Transmembrane Transport Zm00001d043509 Pectinesterase LOC100381490 0042545 Cell Wall Modification 0045490 Pectin Catabolic Process Zm00001d022510 — LOC100194185 0005975 Carbohydrate Metabolic Process Zm00001d014863 — LOC100280442 0006351 Transcription, DNA-templated 0006355 Regulation of Transcription,
DNA-templatedZm00001d027966 — LOC100304379 0010038 Response to Metal Ion 表 5 候选基因GO注释–细胞组分Table 5 GO of the candidate genes-cellular component基因ID 基因说明 基因名称 细胞组分 GO ID GO 注释 Zm00001d010368 Derlin1-1 LOC100037763 0005789 Endoplasmic Reticulum Membrane 0016021 Integral Component of Membrane Zm00001d044606 Grx_S16-Glutaredoxin Subgroup II LOC100193989 0005623 Obsolete Cell Zm00001d011679 High-Affinity Nitrate Transporter 2.3 LOC103636218 0005886 Plasma Membrane 0016021 Integral Component of Membrane 续表 5 基因ID 基因说明 基因名称 细胞组分 GO ID GO 注释 Zm00001d043947 Lipid Phosphate Phosphatase 2 LOC103651387 0016021 Integral Component of Membrane Zm00001d043509 Pectinesterase LOC100381490 0005618 Cell Wall Zm00001d012814 — LOC100274283 0016021 Integral Component of Membrane Zm00001d014863 — LOC100280442 0005634 Nucleus Zm00001d027966 — LOC100304379 0016021 Integral Component of Membrane Zm00001d053563 — LOC100383387 0016021 Integral Component of Membrane Zm00001d024497 — LOC100501068 0016021 Integral Component of Membrane 表 6 候选基因GO注释–分子功能Table 6 GO of the candidate genes-molecular function基因ID 基因说明 基因名称 分子功能 GO ID GO 注释 Zm00001d003086 Aldo-Keto Reductase/ Oxidoreductase LOC100282766 0016491 Oxidoreductase Activity Zm00001d036238 Alpha-L-Fucosidase 2 LOC100193659 0016788 Hydrolase Activity, Acting on Ester Bonds Zm00001d044606 Grx_S16-Glutaredoxin Subgroup Ii LOC100193989 0009055 Electron Transfer Activity 0015035 Protein-Disulfide Reductase Activity Zm00001d043509 Pectinesterase LOC100381490 0030599 Pectinesterase Activity 0045330 Aspartyl Esterase Activity Zm00001d050833 Putative Protein Kinase
Superfamily ProteinLOC100193882 0004713 Protein Tyrosine Kinase Activity 0005524 ATP Binding Zm00001d022510 — LOC100194185 0004134 4-Alpha-Glucanotransferase Activity Zm00001d038651 Jacalin-Related Lectin 3 LOC100272821 0030246 Carbohydrate Binding Zm00001d034662 — LOC100279389 0004674 Protein Serine/Threonine Kinase Activity 0005524 ATP Binding Zm00001d014863 — LOC100280442 0003677 DNA Binding Zm00001d024497 — LOC100501068 0005524 ATP Binding 0016887 ATPase KEGG通路分析结果显示,候选基因主要参与了内质网中的蛋白质加工、氮素代谢、组氨酸代谢、代谢途径、次生代谢产物的生物合成、戊糖和葡萄糖酸酯的相互转化、植物激素信号转导等通路。
详细信息如表7所示。
表 7 候选基因KEGG通路分析Table 7 KEGG pathway of the candidate genes基因ID 基因说明 基因名称 KEGG通路 Zm00001d010368 Derlin1-1 LOC100037763 Protein Processing In Endoplasmic Reticulum Zm00001d011679 High-Affinity Nitrate Transporter 2.3 LOC103636218 Nitrogen Metabolism Zm00001d050694 Serine Decarboxylase 1 LOC103653609 Histidine Metabolism Metabolic Pathways Biosynthesis of Secondary Metabolites Zm00001d043509 Pectinesterase LOC100381490 Pentose and Glucuronate Interconversions Metabolic Pathways Zm00001d004966 BTB/POZ Domain and Ankyrin
Repeat-Containing Protein NH5.1LOC103647110 Plant Hormone Signal Transduction 4. 结束语
在分析了转录组分析方法的进展后,本文提出了融合遗传算法与XGBoost的转录组分析方法。以高质量的玉米转录组测序数据和对应的表型数据作为数据集,研究了影响玉米百粒重性状的相关基因。首先分析了遗传算法和XGBoost用于转录组分析领域的可行性,提出了融合遗传算法和XGBoost的方法——GA-XGBoost。在完成转录组数据的预处理工作和差异表达分析之后,使用本文所提方法对其进行了分析,实验得到了48个与玉米百粒重相关的候选基因,并对其进行了基因本体注释和KEGG通路分析。同时,将本文方法所得模型与分别使用全体基因和差异表达基因进行训练的XGBoost模型进行了比较,在预测结果的准确性上,GA-XGBoost模型具有最低的均方误差,达到3.752,低于使用全体基因的9.183和使用差异表达基因的7.689;在候选基因的范围上,从传统方法直接对154 2个差异表达基因进行分析的基础上缩减到验证48个候选基因,表明本文所提方法能够有效提升对转录组数据的分析能力和效率。
本文虽然实现了对于影响玉米百粒重性状的候选基因挖掘,但仍存在不足之处。融合遗传算法和XGBoost的转录组分析方法,虽然能够得到范围更小的候选基因,但是GA-XGBoost算法本身因为使用遗传算法的原因,导致寻找最优子集时可能会消耗较长时间;XGBoost由于具有较多的参数,而使用网格调参时,随着参数的增多计算时间会大幅增加,如何快速且低消耗地寻找到合适参数使得模型达到最优也是值得探讨的问题。此外,通过功能注释虽然能够在一定程度上表明所选基因与百粒重相关,但还需要进一步构建基因调控网络等步骤切实证明所选基因合理可靠,这将是本课题之后的研究方向。
-
图 1 方法整体流程
Fig. 1 Method overall process
下载:
全尺寸图片
图 2 GA-XGBoost算法
Fig. 2 GA-XGBoost algorithm
下载:
全尺寸图片
图 3 特征编码
Fig. 3 Feature coding
下载:
全尺寸图片
图 4 交叉和突变
Fig. 4 Crossover and mutation
下载:
全尺寸图片
图 5 差异表达基因的MA图
Fig. 5 MA-plot of DEGs
下载:
全尺寸图片
图 6 GA-XGBoost中适应度的变化
Fig. 6 Change of fitness in GA-XGBoost
下载:
全尺寸图片
图 7 XGBoost模型中部分重要特征
Fig. 7 Part of important features in XGBoost model
下载:
全尺寸图片
表 1 差异表达分析结果 (padj < 0.1,419 24个非0基因)
Table 1 Differential expression analysis results (padj < 0.1,419 24 non-zero genes)
差异表达
基因类型Log2FoldChange
(LFC)差异表达
基因数量差异表达
基因占比/%上调基因 LFC > 0 934 2.2 上调基因 LFC > 1 843 2.0 下调基因 LFC < 0 860 2.1 下调基因 LFC < 1 699 1.7 表 2 3种方法的MSE
Table 2 MSE of three methods
方法 MSE 全体基因训练所得XGBoost模型 9.183 差异表达基因所得XGBoost模型 7.689 GA-XGBoost最优子集所得XGBoost模型 3.752 表 3 候选基因数量比较
Table 3 Comparison of the candidate genes number
方法 候选基因数量 差异分析 154 2 GA-XGBoost 48 表 4 候选基因GO注释–生物过程
Table 4 GO of the candidate genes-biological process
基因ID 基因说明 基因名称 生物过程 GO ID GO 注释 Zm00001d044606 Grx_S16-Glutaredoxin Subgroup Ii LOC100193989 0045454 Cell Redox Homeostasis Zm00001d011679 High Affinity Nitrate Transporter 2.5 LOC103636218 0055085 Transmembrane Transport Zm00001d043509 Pectinesterase LOC100381490 0042545 Cell Wall Modification 0045490 Pectin Catabolic Process Zm00001d022510 — LOC100194185 0005975 Carbohydrate Metabolic Process Zm00001d014863 — LOC100280442 0006351 Transcription, DNA-templated 0006355 Regulation of Transcription,
DNA-templatedZm00001d027966 — LOC100304379 0010038 Response to Metal Ion 表 5 候选基因GO注释–细胞组分
Table 5 GO of the candidate genes-cellular component
基因ID 基因说明 基因名称 细胞组分 GO ID GO 注释 Zm00001d010368 Derlin1-1 LOC100037763 0005789 Endoplasmic Reticulum Membrane 0016021 Integral Component of Membrane Zm00001d044606 Grx_S16-Glutaredoxin Subgroup II LOC100193989 0005623 Obsolete Cell Zm00001d011679 High-Affinity Nitrate Transporter 2.3 LOC103636218 0005886 Plasma Membrane 0016021 Integral Component of Membrane 续表 5 基因ID 基因说明 基因名称 细胞组分 GO ID GO 注释 Zm00001d043947 Lipid Phosphate Phosphatase 2 LOC103651387 0016021 Integral Component of Membrane Zm00001d043509 Pectinesterase LOC100381490 0005618 Cell Wall Zm00001d012814 — LOC100274283 0016021 Integral Component of Membrane Zm00001d014863 — LOC100280442 0005634 Nucleus Zm00001d027966 — LOC100304379 0016021 Integral Component of Membrane Zm00001d053563 — LOC100383387 0016021 Integral Component of Membrane Zm00001d024497 — LOC100501068 0016021 Integral Component of Membrane 表 6 候选基因GO注释–分子功能
Table 6 GO of the candidate genes-molecular function
基因ID 基因说明 基因名称 分子功能 GO ID GO 注释 Zm00001d003086 Aldo-Keto Reductase/ Oxidoreductase LOC100282766 0016491 Oxidoreductase Activity Zm00001d036238 Alpha-L-Fucosidase 2 LOC100193659 0016788 Hydrolase Activity, Acting on Ester Bonds Zm00001d044606 Grx_S16-Glutaredoxin Subgroup Ii LOC100193989 0009055 Electron Transfer Activity 0015035 Protein-Disulfide Reductase Activity Zm00001d043509 Pectinesterase LOC100381490 0030599 Pectinesterase Activity 0045330 Aspartyl Esterase Activity Zm00001d050833 Putative Protein Kinase
Superfamily ProteinLOC100193882 0004713 Protein Tyrosine Kinase Activity 0005524 ATP Binding Zm00001d022510 — LOC100194185 0004134 4-Alpha-Glucanotransferase Activity Zm00001d038651 Jacalin-Related Lectin 3 LOC100272821 0030246 Carbohydrate Binding Zm00001d034662 — LOC100279389 0004674 Protein Serine/Threonine Kinase Activity 0005524 ATP Binding Zm00001d014863 — LOC100280442 0003677 DNA Binding Zm00001d024497 — LOC100501068 0005524 ATP Binding 0016887 ATPase 表 7 候选基因KEGG通路分析
Table 7 KEGG pathway of the candidate genes
基因ID 基因说明 基因名称 KEGG通路 Zm00001d010368 Derlin1-1 LOC100037763 Protein Processing In Endoplasmic Reticulum Zm00001d011679 High-Affinity Nitrate Transporter 2.3 LOC103636218 Nitrogen Metabolism Zm00001d050694 Serine Decarboxylase 1 LOC103653609 Histidine Metabolism Metabolic Pathways Biosynthesis of Secondary Metabolites Zm00001d043509 Pectinesterase LOC100381490 Pentose and Glucuronate Interconversions Metabolic Pathways Zm00001d004966 BTB/POZ Domain and Ankyrin
Repeat-Containing Protein NH5.1LOC103647110 Plant Hormone Signal Transduction -
[1] MOROZOVA O, HIRST M, MARRA M A. Applications of new sequencing technologies for transcriptome analysis[J]. Annual review of genomics and human genetics, 2009, 10: 135–151. [2] 郭茂祖, 杨帅, 赵玲玲. 基于RNA-Seq的转录组分析方法[J]. 计算机科学, 2020, 47(11A): 35–39.GUO Maozu, YANG Shuai, ZHAO Lingling. Transcriptome analysis method based on RNA-Seq[J]. Computer science, 2020, 47(11A): 35–39. [3] KUKURBA K R, MONTGOMERY S B. RNA sequencing and analysis[J]. Cold spring harbor protocols, 2015, 2015(11): 951–961. [4] XU Maoqi, CHEN Liang. An empirical likelihood ratio test robust to individual heterogeneity for differential expression analysis of RNA-seq[J]. Briefings in bioinformatics, 2018, 19(1): 109–117. [5] ZHAO Xin, DOU Jian, CAO Jinglin, et al. Uncovering the potential differentially expressed miRNAs as diagnostic biomarkers for hepatocellular carcinoma based on machine learning in The Cancer Genome Atlas database[J]. Oncology reports, 2020, 43(6): 1771–1784. [6] 白云帆. 基于网络分析和机器学习的肝癌中糖链相关基因筛选[D]. 哈尔滨: 哈尔滨工业大学, 2019.BAI Yunfan. Screening of sugar chain related genes in hepatocellular carcinoma based on network analysis and machine learning[D]. Harbin: Harbin Institute of Technology, 2019. [7] SANTOS C A, ANDRADE S C S, TEIXEIRA A K, et al. Transcriptome differential expression analysis reveals the activated genes in Litopenaeus vannamei shrimp families of superior growth performance[J]. Aquaculture, 2021, 531: 735871. [8] ABBAS S Z, QADIR M I, MUHAMMAD S A. Systems-level differential gene expression analysis reveals new genetic variants of oral cancer[J]. Scientific reports, 2020, 10(1): 14667. [9] HOLLAND J. Adaptation in natural and artificial systems: an introductory analysis with application to biology [J]. Control & Artificial Intelligence, 1975:1–198. [10] XING Xuguang, LIU Ye, GARG A, et al. An improved genetic algorithm for determining modified water-retention model for biochar-amended soil[J]. CATENA, 2021, 200: 105143. [11] REVUELTA E C, CHÁVEZ M J, VERA J A B, et al. Optimization of laser scanner positioning networks for architectural surveys through the design of genetic algorithms[J]. Measurement, 2021, 174: 108898. [12] LIU Kunhong, JIN Qiushi, XU Huangchao, et al. Micro-expression recognition using advanced genetic algorithm[J]. Signal processing: image communication, 2021, 93: 116153. [13] XUE Jingjing, AHMADIAN R, JONES O, et al. Design of tidal range energy generation schemes using a Genetic Algorithm model[J]. Applied energy, 2021, 286: 116506. [14] YARSYARSKY P. Using a genetic algorithm to fit parameters of a COVID-19 SEIR model for US states[J]. Mathematics and computers in simulation, 2021, 185: 687–695. [15] PENG Cheng, WU Xinyu, YUAN Wen, et al. MGRFE: multilayer recursive feature elimination based on an embedded genetic algorithm for cancer classification[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2021, 18(2): 621–632. [16] AKARSU H, AGUILAR-BULTET L, FALQUET L. deltaRpkm: an R package for a rapid detection of differential gene presence between related bacterial genomes[J]. BMC bioinformatics, 2019, 20(1): 621. [17] KUMAR S, KALRA S, SINGH B, et al. RNA-Seq mediated root transcriptome analysis of Chlorophytum borivilianum for identification of genes involved in saponin biosynthesis[J]. Functional & integrative genomics, 2016, 16(1): 37–55. [18] SUN Qiuli, ZHAO Chunpeng, WANG Tianyun, et al. Expression profile analysis of long non-coding RNA associated with vincristine resistance in colon cancer cells by next-generation sequencing[J]. Gene, 2015, 572(1): 79–86. [19] MA Wentai, LIU Zhaoyu, CHEN Xiaozhou, et al. A protein identification algorithm for tandem mass spectrometry by incorporating the abundance of mRNA into a binomial probability scoring model[J]. Journal of proteomics, 2019, 197: 53–59. [20] CHEN Tianwu, GUESTRIN C. XGBoost: a scalable tree boosting system[C]//Proceedings of the 22nd ACM SIGKDD International Conference. San Francisco, USA, 2016: 785–794. [21] JIANG Yiquan, CAO Su’e, CAO Shilei, et al. Preoperative identification of microvascular invasion in hepatocellular carcinoma by XGBoost and deep learning[J]. Journal of cancer research and clinical oncology, 2021, 147(3): 821–833. [22] ABDU-ALJABAR R D, AWAD O A. A Comparative analysis study of lung cancer detection and relapse prediction using XGBoost classifier[J]. IOP conference series: materials science and engineering, 2021, 1076: 012048. [23] CHEN Sijie, ZHOU Wenjing, TU Jinghui, et al. A novel XGBoost method to infer the primary lesion of 20 solid tumor types from gene expression data[J]. Frontiers in genetics, 2021, 12: 632761. [24] ZHAO Xiaowei, ZHANG Ye, NING Qiao, et al. Identifying N6-methyladenosine sites using extreme gradient boosting system optimized by particle swarm optimizer[J]. Journal of theoretical biology, 2019, 467: 39–47. [25] GUYON I, WESTON J, BARNHILL S, et al. Gene selection for cancer classification using support vector machines[J]. Machine learning, 2002, 46(1/2/3): 389–422.




















































































