 2019, Vol. 40
2019, Vol. 40文章信息
- 朱猛, 吕筠, 余灿清, 靳光付, 郭彧, 卞铮, Walters Robin, Millwood Iona, 陈铮鸣, 沈洪兵, 胡志斌, 李立明, 代表中国慢性病前瞻性研究项目协作组.
- Zhu Meng, Lyu Jun, Yu Canqing, Jin Guangfu, Guo Yu, Bian Zheng, Walters Robin, Millwood Iona, Chen Zhengming, Shen Hongbing, Hu Zhibin, Li Liming, for the China Kadoorie Biobank Collaborative Group.
- 中国不同地区群体遗传结构差异及调整策略研究
- Study on genetic structure differences and adjustment strategies in different areas of China
- 中华流行病学杂志, 2019, 40(1): 20-25
- Chinese Journal of Epidemiology, 2019, 40(1): 20-25
- http://dx.doi.org/10.3760/cma.j.issn.0254-6450.2019.01.006
-
文章历史
收稿日期: 2018-08-09




2. 北京大学公共卫生学院流行病与卫生统计学系 100191;
3. 北京大学分子心血管学教育部重点实验室 100191;
4. 中国医学科学院, 北京 100730;
5. 英国牛津大学临床与流行病学研究中心纳菲尔德人群健康系 OX3 7LF
2. Department of Epidemiology and Biostatistics, School of Public Health, Peking University, Beijing 100191, China;
3. Key Laboratory of Molecular Cardiovascular Sciences, Ministry of Education, Peking University, Beijing 100191, China;
4. Chinese Academy of Medical Sciences, Beijing 100730, China;
5. Clinical Trial Service Unit and Epidemiological Studies Unit(CTSU), Nuffield Department of Population Health, University of Oxford, Oxford OX3 7LF, UK
近年来,随着高通量分型技术平台的建立和完善,全基因组关联研究(Genome-wide association studies,GWAS)已经成为分子流行病学研究中探索复杂疾病遗传易感性的有力工具[1]。GWAS通过在全基因组范围内同时检测几十万甚至上百万个单核苷酸多态性(Single nucleotide polymorphism,SNP)位点,利用大样本筛选结合多阶段验证,最终确定与疾病或性状相关的SNP位点。基于中国人群的GWAS也取得了丰硕的研究成果,截至2018年7月,GWAS Catalog已经收录了460余项基于中国人群的GWAS,报道了4 200多个与不同性状相关的遗传易感位点[2]。
在GWAS等分子流行病学研究中,群体遗传结构是重要的混杂因素之一。群体遗传结构是指来自不同组别、不同区域的个体在进化选择中存在遗传差异,导致不同亚群间同一等位基因频率存在显著差异,表现为群体分层和亲缘关系。当GWAS包含不同遗传结构的样本时,这种亚群间的频率差异可能被解读为表型差异,造成假阳性关联结果。在GWAS分析中,我们一般可以通过基因组控制、分层分析、主成分分析或混合线性模型来控制群体遗传结构对关联结果的影响[3]。
《“十三五”国家科技创新规划》围绕慢性非传染性疾病、精准医学等部署建立了多项国家级自然人群大队列和慢病队列,为疾病的遗传病因研究提供了新的契机。然而,由于大队列涉及地区广,队列成员遗传结构复杂,而且样本间存在错综复杂的亲缘关系,增加了群体遗传结构的复杂性,传统的分析策略难以满足要求。为了系统评价大队列研究中群体遗传结构的影响,本研究以中国慢性病前瞻性(China Kadoorie Biobank,CKB)队列GWAS数据为基础,系统描述我国不同地区样本的人群遗传结构特征,探索并评价不同分析方案控制队列样本群体遗传结构混杂因素的效果。
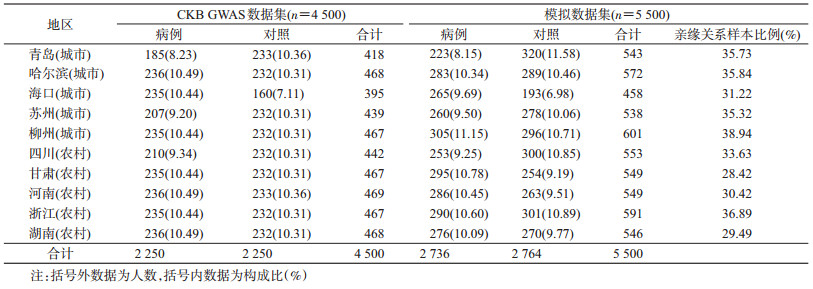
对象与方法1.研究对象:CKB项目于2004-2008年在中国10个项目地区(5个城市地区:山东省青岛市李沧区、黑龙江省哈尔滨市南岗区、海南省海口市美兰区、江苏省苏州市吴中区、广西壮族自治区柳州市;5个农村地区:四川省彭州市、甘肃省天水市麦积区、河南省辉县市、浙江省桐乡市、湖南省浏阳市)招募调查对象并完成基线调查。为简化表述且方便识别,以城市名称表示城市项目点,以省份名称表示农村项目点,有关项目的介绍详见文献[4-5]。本研究共纳入10个项目地区4 500例样本的全基因组分型数据,包含2 250例脑卒中病例与2 250例匹配地区、年龄、性别的健康对照。
2.研究内容与方案:本研究根据CKB队列的4 500例GWAS数据进行两部分研究内容。一是描述我国不同地区人群的遗传结构与地理分布的特征,二是生成亲缘关系更为复杂的模拟数据集。在此基础上,比较6种不同分析策略控制群体遗传结构混杂的效果,包括:①传统logistic回归分析;②logistic回归调整人群遗传结构主成分;③根据地区进行亚组分析,并使用Meta分析合并;④使用混合线性模型,引入样本亲属关系矩阵作为随机效应变量进行关联分析;⑤在混合线性模型的基础上,进一步调整人群遗传结构PCs;⑥在每个地区使用混合线性模型进行关联分析,最后使用Meta分析合并。
3. GWAS数据集:本研究使用的4 500例GWAS原始数据来源于CKB队列的10个项目点。所有样本均使用自主设计的Affymetrix Axiom® CKB array分型,芯片共包含78余万个位点。经过系统质量控制后(剔除分型成功率<95%,哈代-温伯格平衡P<1.0×10-4,最小等位基因频率MAF<0.01),本研究共纳入527 260个位点进行后续分析。样本质控未发现不合格样本。为了模拟大队列数据的复杂亲缘关系特征,我们在4 500例样本数据的基础上,随机抽取1 000例样本生成与其存在二级亲缘关系的模拟数据集(0.4>PI_HAT>0.25),并与原始的4 500例样本合并,产生包含5 500例样本的模拟数据集,用于不同分析策略的评价。
4.膨胀因子(inflation factor,λ):λ是Devlin和Roeder[6]最早提出的衡量群体结构对关联分析影响的指标。在进行GWAS关联分析时,λ服从χ2分布,大小由群体结构控制,同时也受样本量的影响。假设从基因组随机选取一组无关的SNP位点,则每个位点的检验统计量服从χ2分布,期望为λ,而原假设为随机变量的统计量服从自由度为1的χ2分布,期望为1。实际研究中多采用M进行比较,计算公式为
其中,0.454 9为原假设下统计量的中位数,M(Ti2)为实际研究统计量的中位数。一般认为,当λ<1.01时,群体结构对关联结果影响很小;当1.01<λ<1.05时,群体结构的影响中等,但在接受的范围之内;当λ>1.1时,群体结构对关联结果影响很大[3, 6]。
5.统计学分析:群体遗传结构主成分分析基于123 122个无连锁不平衡(r2<0.1)的SNP位点,分析使用EIGENSTRAT软件的smartpca程序包,基于无亲缘关系的样本(PI_HAT<0.125)计算主成分,并投射到所有样本[7]。当合并分析时,根据总体样本计算主成分;当分地区进行亚组分析时,根据各地区样本计算区域内主成分。在总体样本中,根据计算产生的第一、第二主成分绘制CKB队列不同地区样本的二维结构图。SNP位点的logistic回归分析使用PLINK软件[8],全基因组Meta分析使用GWAMA软件[9],混合线性模型分析使用R软件包GMMAT[10]。研究调整变量为年龄、性别及前10个PCs。使用R软件包GenABEL估算关联结果的λ,并绘制Q-Q图[11]。
结果1.一般情况:本研究纳入的4 500例CKB队列样本地区分布情况见表 1,各项目地区样本量在395~469例,病例组与对照组占比均在10%左右。模拟数据集各地区样本比例与CKB原始数据基本一致,存在亲缘关系(PI_HAT≥0.25)样本总体比例为33.71%,各地区分布在28.42%~38.94%之间(表 1)。
2.人群遗传结构与地区分布特征及其影响:在GWAS中,主成分分析根据全基因组等位基因构建关系矩阵,然后计算关系矩阵的特征值和特征向量,根据特征值的大小选择少数几个特征向量代替关系矩阵。本研究中选取主成分分析的第一、二主成分代表样本的人群结构分布。根据10个项目地区4 500例样本的GWAS数据集进行主成分分析,并提取第一、二主成分绘制人群遗传结构主成分二维图。如图 1所示,我们观察到不同地区样本存在显著的遗传结构差异,除了哈尔滨地区样本以外,其他地区人群遗传结构主成分分布与项目地区的地理分布基本一致,第一主成分对应不同地区的纬度,第二主成分对应不同地区的经度。不同地区来源的样本基本可以通过第一、二主成分有效区分,说明人群遗传结构在我国普遍存在地区差异。而哈尔滨地区样本人群遗传结构与青岛地区极为相近,提示两组人群具有相近的亚群起源。

|
| 图 1 CKB队列项目点人群遗传结构特征 |
上述结果表明我国人口遗传结构的复杂性,也进一步确证了在GWAS中根据样本地区来源进行匹配的必要性。尽管本研究纳入的CKB队列样本每个地区病例、对照样本所占比例极为相近,但当不调整人群遗传结构PCs时,λ仍达到了1.10;而进一步调整PCs后,λ降为1.00,证实了调整遗传结构PCs是控制人群遗传结构影响的有效手段[9]。
3.不同分析策略控制群体遗传结构影响的效果评价:除了遗传结构的地区差异,大队列遗传数据的另一个特点是存在广泛的亲缘关系。如果直接删除存在亲缘关系的样本,会损失大量研究信息,导致研究效率降低。以生成的5 500例模拟数据为例,不调整遗传结构PCs时,λ为1.16(图 2A);而调整遗传结构PCs后,λ降为1.12(图 2B);即使根据样本的地区来源进行亚组分析,并调整区域内的遗传结构PCs,最后使用全基因组Meta分析进行关联结果合并,λ仍达到了1.08(图 2C),均高于目前一般可接受的阈值1.05。

|
| 注:A.传统logistic回归分析;B. logistic回归调整人群遗传结构PCs;C.根据地区进行亚组分析,并使用Meta合并;D.使用混合线性模型,引入样本关系矩阵作为协变量进行关联分析;E.在混合线性模型的基础上,进一步调整人群遗传结构PCs;F.在每个地区使用混合线性模型进行关联分析,最后使用Meta分析合并 图 2 不同分析策略控制群体遗传结构影响的效果评价 |
为了进一步排除样本间亲缘关系的影响,我们使用混合线性模型引入亲属关系(kinship)矩阵作为随机效应量进行关联分析,结果显示人群遗传结构影响得到有效控制,λ=0.99(图 2D);在引入亲属关系矩阵的基础上,进一步调整人群遗传结构PCs,λ=0.99(图 2E);在10个项目地区分别使用混合线性模型进行关联分析,同时调整区域内人群遗传结构PCs,最后进行Meta分析,λ=0.99(图 2F)。上述研究结果表明,针对既存在群体遗传结构差异又存在大量亲属关系的大队列遗传数据进行分析时,使用混合线性模型可以有效控制群体遗传结构的影响。
讨论在过去10年间,GWAS在复杂疾病的遗传病因领域取得了一系列重要发现,推动了分子流行病学研究的快速发展。随着我国多项大规模前瞻性队列的启动,分子流行病学研究将开启新的纪元[12]。在此时期,系统探讨我国大规模队列遗传数据的特征,并探索适用的分析策略,必要且及时。本研究基于CKB队列GWAS数据,系统描述了我国人群的群体遗传结构特征,发现第一、二主成分与样本所在地区的纬度、经度分布一致;针对大队列遗传数据群体遗传结构复杂、亲属关系广泛等特征,系统比较了多种不同分析策略,发现使用混合线性模型调整亲属关系可以有效控制群体遗传结构的影响。
Chen等[13]曾利用GWAS数据对来自我国10个省份的6 000余名汉族人群进行了遗传结构分析,研究发现我国南-北方人群遗传结构存在显著差异,模拟实验表明当GWAS中病例组来源于北方人群,而对照组包含20%~100%的南方人群时,λ可达1.17~5.49,远远高于可接受的阈值1.05。而我们的研究发现,我国人口遗传结构除了存在南-北差异(第一主成分)外,也呈现东-西分布差异,根据绘制的二维遗传结构图,我们发现在相似纬度的柳州-海口、四川-湖南、甘肃-河南-青岛等地区人口的第二主成分也呈现明显差异,且与地理分布一致。而哈尔滨人群的遗传特征与青岛地区极为相近,这与我国历史上著名的一次人口大迁徙“闯关东”相符,提示哈尔滨与青岛地区人群可能来源于相同亚群[14]。尽管我国人口结构复杂,但在我们的数据中,当研究样本病例与对照分布比例接近,同时调整人群遗传结构主成分时,仍可以有效控制群体遗传结构的影响。
当样本既存在明显的群体结构差异,同时又存在广泛的亲属关系时,传统分析方法均存在明显缺陷。当引入亲属关系矩阵作为随机效应量进行关联分析时,无论是否进一步调整人群遗传结构主成分,关联λ均得到明显控制,且与更为严格的分地区计算Meta合并相比,λ无明显差异。然而,我们观察到分地区计算Meta合并策略的检验效能低于合并分析(图 2E vs. 图 2F)。尽管本研究中,在混合线性模型的基础上进一步调整人群遗传结构主成分并没有影响λ,但是目前多数研究仍推荐调整人群遗传结构主成分[15-16],该策略也是目前UK Biobank等国外大型队列研究最常用的分析方法[17]。在我们的模拟分析中,两者关联结果基本一致(r=0.98,图 2D vs. 图 2E)。
目前支持GWAS数据混合线性模型分析的软件主要包括BOLT-LMM、GEMMA、GMMAT以及SAIGE,其中前两者主要适用于连续型变量表型,后两者适用于病例对照研究[11, 18-20]。不同的软件拥有各自的特点,相较于GEMMA与GMMAT,BOLT- LMM与SAIGE更适用研究样本较大的关联分析,但是SAIGE目前并不支持存在基因型缺失的数据集。本研究选择GMMAT软件,主要因为本研究使用的是原始分型数据,并未进行基因型填补,存在部分的基因型信息缺失。此外,针对大队列研究中的罕见疾病,病例/对照比常呈现不均衡特征(病例:对照<1 : 10),此时GMMAT方法的Ⅰ类错误率会大大增加,SAIGE更有优势[20]。
本研究通过分析CKB项目的部分GWAS数据,描述了我国不同地区人群遗传结构的基本特征;通过模拟分析,评价了多种分析策略控制群体遗传结构的效果。本研究的开展,对于理解分子流行病学研究中群体遗传结构的影响,选择适合大队列复杂遗传结构数据特征的分析策略提供了宝贵经验。
利益冲突 所有作者均声明不存在利益冲突
志谢 感谢所有CKB项目的成员和现场调查员;感谢项目管理委员会、国家项目办公室、牛津协作中心和10个项目地区办公室的所有工作人员
| [1] |
Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery:biology, function, and translation[J]. Am J Hum Genet, 2017, 101(1): 15-22. DOI:10.1016/j.ajhg.2017.06.005 |
| [2] |
MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog)[J]. Nucleic Acids Res, 2017, 45(D1): D896-901. DOI:10.1093/nar/gkw1133 |
| [3] |
郝兴杰, 胡林, 张淑君. 全基因组关联分析方法的研究进展[J]. 兽牧兽医学报, 2016, 47(2): 213-217. Hao XJ, Hu L, Zhang SJ. Progresses in research of genome-wide association study methods[J]. Acta Vet Zootech Sin, 2016, 47(2): 213-217. DOI:10.11843/j.issn.0366-6964.2016.02.001 |
| [4] |
Chen ZM, Lee L, Chen JS, et al. Cohort profile:the Kadoorie study of chronic disease in China (KSCDC)[J]. Int J Epidemiol, 2005, 34(6): 1243-1249. DOI:10.1093/ije/dyi174 |
| [5] |
李立明, 吕筠, 郭彧, 等. 中国慢性病前瞻性研究:研究方法和调查对象的基线特征[J]. 中华流行病学杂志, 2012, 33(3): 249-255. Li LM, Lyu J, Guo Y, et al. The China Kadoorie Biobank:related methodology and baseline characteristics of the participants[J]. Chin J Epidemiol, 2012, 33(3): 249-255. DOI:10.3760/cma.j.issn.0254-6450.2012.03.001 |
| [6] |
Devlin B, Roeder K. Genomic control for association studies[J]. Biometrics, 1999, 55(4): 997-1004. DOI:10.1111/j.0006-341X.1999.00997.x |
| [7] |
Purcell S, Neale B, Todd-Brown K, et al. PLINK:a tool set for whole-genome association and population-based linkage analyses[J]. Am J Hum Genet, 2007, 81(3): 559-575. DOI:10.1086/519795 |
| [8] |
Mägi R, Morris AP. GWAMA:software for genome-wide association Meta-analysis[J]. BMC Bioinformatics, 2010, 11: 288. DOI:10.1186/1471-2105-11-288 |
| [9] |
Chen H, Wang CL, Conomos MP, et al. Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models[J]. Am J Hum Genet, 2016, 98(4): 653-666. DOI:10.1016/j.ajhg.2016.02.012 |
| [10] |
Aulchenko YS, Ripke S, Isaacs A, et al. GenABEL:an R library for genome-wide association analysis[J]. Bioinformatics, 2007, 23(10): 1294-1296. DOI:10.1093/bioinformatics/btm108 |
| [11] |
Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies[J]. Nat Genet, 2006, 38(8): 904-909. DOI:10.1038/ng1847 |
| [12] |
Wijmenga C, Zhernakova A. The importance of cohort studies in the post-GWAS era[J]. Nat Genet, 2018, 50(3): 322-328. DOI:10.1038/s41588-018-0066-3 |
| [13] |
Chen JM, Zheng HF, Bei JX, et al. Genetic structure of the Han Chinese population revealed by genome-wide SNP variation[J]. Am J Hum Genet, 2009, 85(6): 775-785. DOI:10.1016/j.ajhg.2009.10.016 |
| [14] |
范立君, 谭玉秀. 百年来国内清代东北移民史研究述评[J]. 中国史研究动态, 2013(5): 64-72. Fan LJ, Tan YX. Review of history of the Northeast immigrants in Qing dynasty during the past century[J]. Research Trends of Chin History, 2013(5): 64-72. |
| [15] |
Weir BS, Goudet J. A unified characterization of population structure and relatedness[J]. Genetics, 2017, 206(4): 2085-2103. DOI:10.1534/genetics.116.198424 |
| [16] |
Hoffman GE. Correcting for population structure and kinship using the linear mixed model:theory and extensions[J]. PLoS One, 2013, 8(10): e75707. DOI:10.1371/journal.pone.0075707 |
| [17] |
Davies G, Marioni RE, Liewald DC, et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112151)[J]. Mol Psych, 2016, 21(6): 758-767. DOI:10.1038/mp.2016.45 |
| [18] |
Loh PR, Tucker G, Bulik-Sullivan BK, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts[J]. Nat Genet, 2015, 47(3): 284-290. DOI:10.1038/ng.3190 |
| [19] |
Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies[J]. Nat Genet, 2012, 44(7): 821-824. DOI:10.1038/ng.2310 |
| [20] |
Zhou W, Nielsen JB, Fritsche LG, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies[J]. bioRxiv, 2017, 212357. DOI:10.1101/212357 |