 2026, Vol. 37
2026, Vol. 37 


b National Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, China
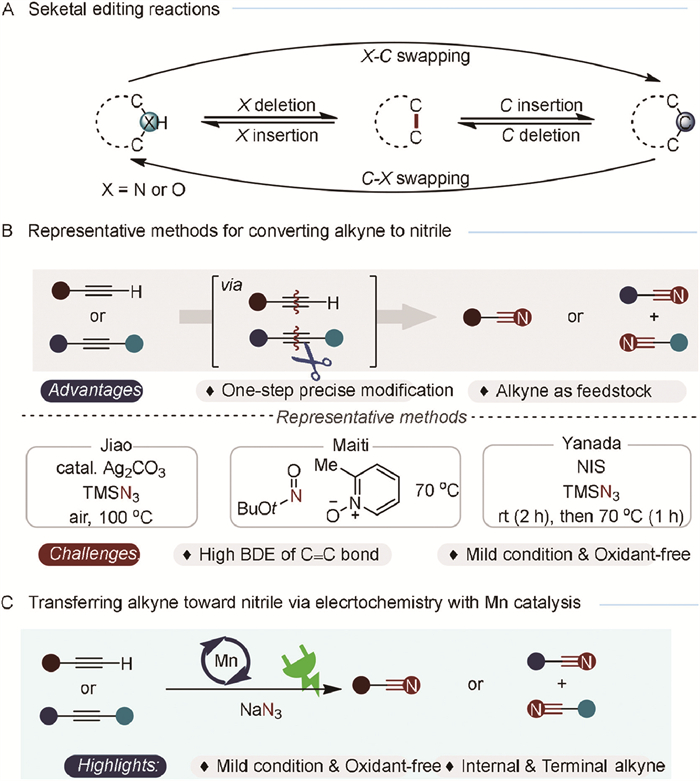
Developing efficient and chemoselective methodologies for crafting complex molecules remains a central challenge in organic synthesis and drug discovery, especially for advancing the chemical sciences [1-3]. Significant progress has been made in late-stage functionalization and molecular editing, particularly in skeletal editing involving carbon and nitrogen atoms [4-9]. Atom substitution within a molecule can have profound effects, making it a powerful tool for constructing intricate compounds (Scheme 1A). Recent research has focused on transformations of C—H and C=C bonds, with particular emphasis on C—H functionalization [10-18] and the oxidative cleavage of C=C bonds [19-24] to generate carbonyl moieties. However, the direct manipulation of alkynes [25], remains less developed which are hindered by the high bond dissociation energy (>200 kcal/mol for C≡C bonds). Given the ubiquitous structural motifs and importance in synthesizing natural products, pharmaceuticals, and materials, exploring methodologies for the precise modification of alkynes is of great interest [26]. Thus, exploring precision modification techniques for alkynes is inherently intriguing [27-30]. Extensive research by the Jiao [31], Yanada [32], Maiti [33], and Song [34] groups has focused on utilizing alkynes to construct nitriles, which are highly valued for their versatile synthetic applications in natural products, pharmaceuticals, agrochemicals, materials, and dyes (Scheme 1B) [35-40]. However, the high energy barriers associated with carbon-carbon triple bonds mean that current methods for cleaving these bonds to produce nitriles often require harsh conditions and stoichiometric oxidants.

|
Download:
|
| Scheme 1. Molecular editing and incorporation reactions. (A) Seketal editing reactions. (B) Oxidation methods for converting alkyne to nitrile. (C) Electrochemical Mn-catalyzed nitrogenation of alkynes to nitriles. | |
The unique features of electrosynthesis—scalability, con-trollability, functional group umpolung, and environmental friendliness—enable electrochemistry to effectively address traditional challenges [41-47]. Through single-electron transfers between the electrode and substrate molecule, electrochemistry has emerged as a powerful method for generating radical species [48-50]. Building on the electro-manganese co-catalyzed azidation of hydrocarbons via a radical process [51-56], we propose an electrochemical strategy for the nitrogenation of alkynes to nitriles via C≡C bond cleavage (Scheme 1C). The manganese catalyst facilitates the generation of azide radicals, accelerating the diazidation of alkynes [57,58]. Subsequently, the rapid departure of nitrogen promotes the rearrangement of intermediates, culminating in the formation of nitriles [59-63]. This protocol demonstrates extensive functional-group tolerance and eliminates the need for stoichiometric chemical oxidants in the nitrogenation of alkynes to nitriles.
We began our investigations using phenylacetylene (1a) as the model substrate and NaN3 as the nitrogen source (Table 1). Employing 5 mol% Mn(acac)3 as the catalyst and 10 mol% 1,10-Phen as the ligand, we were gratified to achieve the desired product, benzonitrile (2a), with a commendable yield of 78% under ambient conditions. The reaction was con-ducted at room temperature with a constant current of 8 mA in an undivided cell, utilizing a carbon rod anode and a platinum plate cathode (entry 1). Exploring the effect of current intensity revealed that deviations from the optimized 8 mA significantly reduced yields (entries 2 and 3). Substituting Mn(acac)3 with manganese(Ⅱ) salts or other transition metals such as CuI and AgNO3 markedly diminished the reactivity, underscoring the unique efficacy of Mn(acac)3 in this transformation (entries 4–6). The absence of 1,10-Phen led to a dramatic decrease in yield to 22%, emphasizing its critical role in enhancing the manganese catalyst's performance (entry 7). Furthermore, the selection of the nitrogen source and electrode material proved vital for achieving optimal reactivity (entries 8 and 9). Control experiments confirmed that the manganese catalyst, NaN3, 1,10-phenanthroline, and constant current are all essential for the successful execution of the reaction (entries 10–13).
|
|
Table 1 Optimization of the reaction conditions.a |

{kind=link}
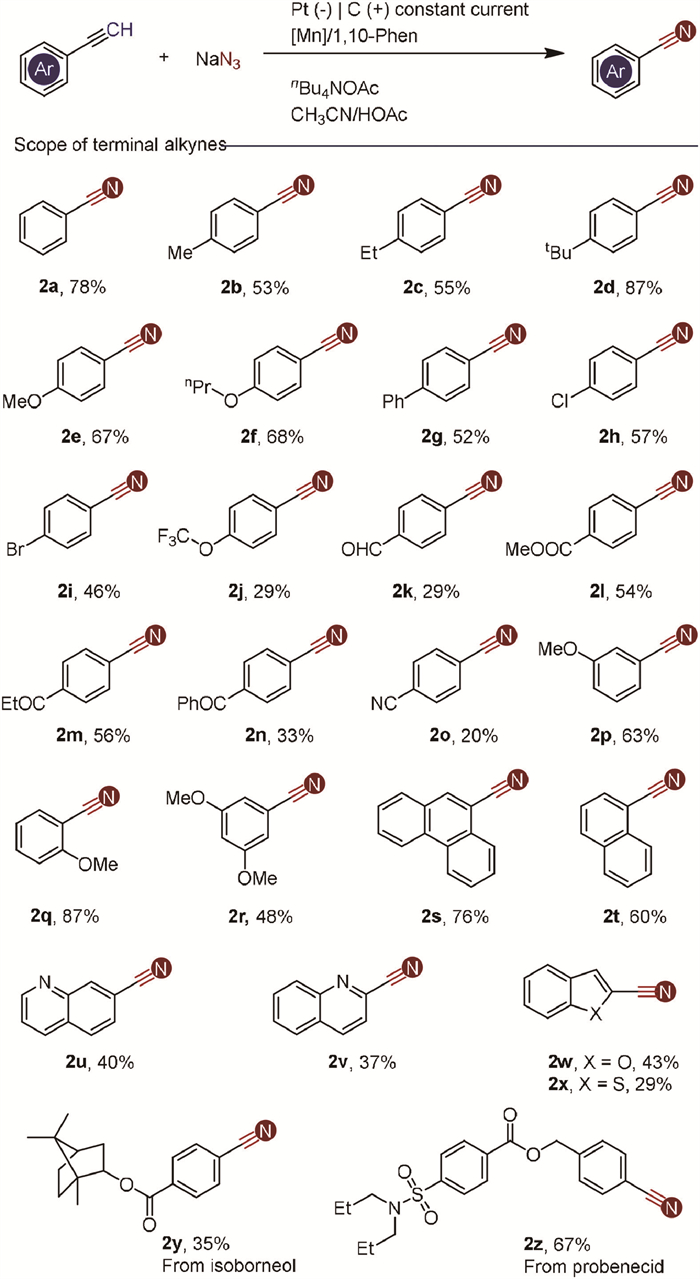
With optimized conditions established, we explored a diverse set of aryl terminal alkynes (Scheme 2). Arenes substituted with electron-donating groups such as methyl (Me), ethyl (Et), tert-butyl (tBu), methoxy (MeO), and propoxy (nPrO) proved effective, yielding the desired nitrile products in good yields (2b-2f). Halogenated arenes, including those with chlorine (Cl) and bromine (Br), were also tolerated (2h and 2i), providing opportunities for downstream synthetic transformations. Various functional groups, including trifluoromethoxy, ketone, aldehyde, ester, and cyano moieties, were compatible under the reaction conditions (2j-2o), though some yields were slightly lower. Substrates with one or more methoxy groups at various positions on the aromatic ring reacted smoothly, demonstrating the robustness of the reaction (2p-2r). The relatively lower yields observed with phenylacetylene derivatives may be attributed to inductive effects. Polycyclic aromatic acetylenes and 4-biphenylacetylene were also suitable, efficiently yielding the corresponding nitriles (2s and 2t). Terminal alkynes containing heterocyclic moieties such as quinoline, benzofuran, and benzothiophene were successfully converted to the desired nitrile compounds (2u-2x). Furthermore, the reaction enabled the functionalization of complex molecules like dl-isoborneol and probenecid-containing alkynes (2y and 2z), demonstrating the strategy's potential applicability in synthesizing pharmaceutical intermediates.

|
Download:
|
| Scheme 2. Substrate scope of terminal alkynes. Reaction conditions: alkynes (1.0 mmol), NaN3 (5.0 mmol), Mn(acac)3 (0.05 mmol), 1,10-Phen (0.1 mmol), and nBu4NOAc in CH3CN (8.5 mL)/HOAc (1.5 mL); stirring under nitrogen atmosphere was electrolyzed at a constant current of 8.0 mA at 25 ℃ for 22 h. | |
{kind=link}
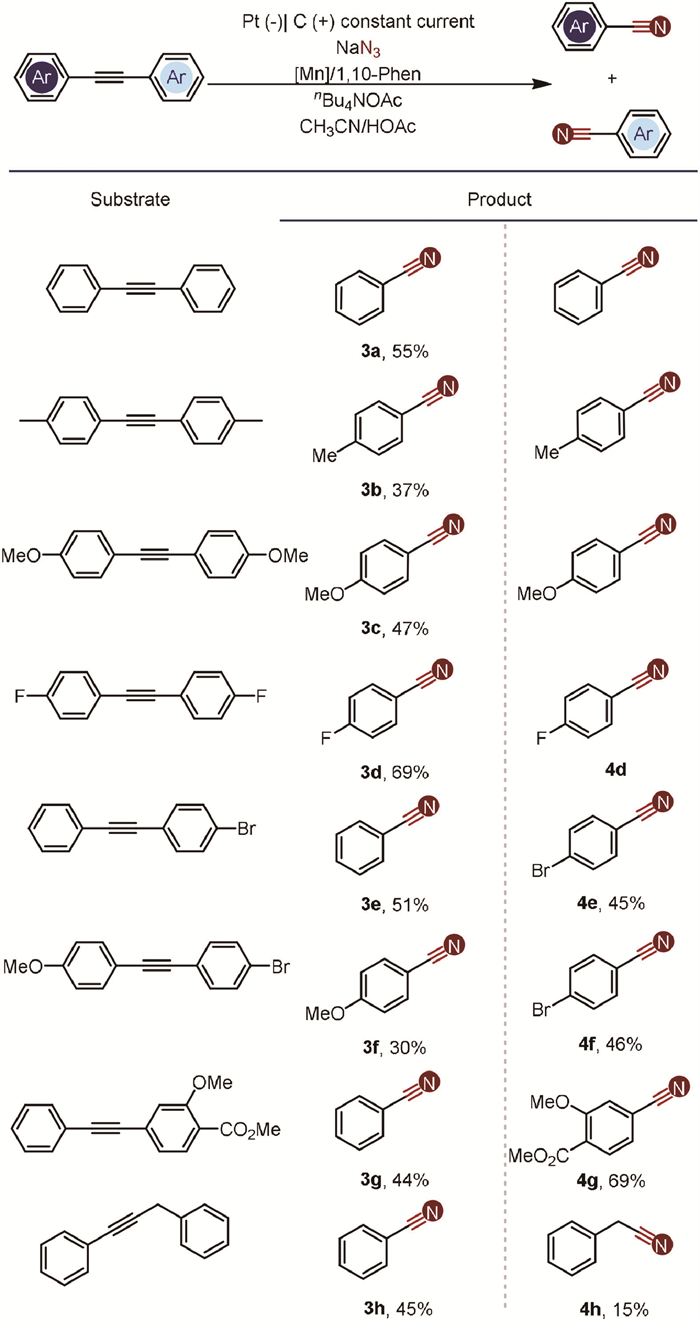
The scope of internal alkynes was further examined, as il-lustrated in Scheme 3. Our system demonstrated compatibility with a variety of functional groups, including methyl, methoxy, and halogens, confirming its versatility. Symmetrical internal alkynes yielded the desired nitrile products with moderate efficiency (3a-3d), indicating the method's reliability under these conditions. Unsymmetrical internal alkynes also proved suitable for this transformation, resulting in the formation of two distinct nitriles with moderate yields (3e-3g). However, the yield will be greatly reduced, when using internal alkynes with alkyl as the ingredient (3h and 4h). And when the aromatic rings on both sides contain strong electron-withdrawing groups, the reaction proceeds with difficulty.

|
Download:
|
| Scheme 3. Substrate scope of internal alkynes. Internal alkynes (0.5 mmol), NaN3 (5.0 mmol), Mn(acac)3 (0.1 mmol), 1,10-Phen (0.1 mmol), and nBu4NOAc in CH3CN (5.5 mL)/HOAc (1.5 mL); stirring under nitrogen atmosphere was electrolyzed at a constant current of 8.0 mA at 25 ℃ for 15 h. | |
{kind=link}
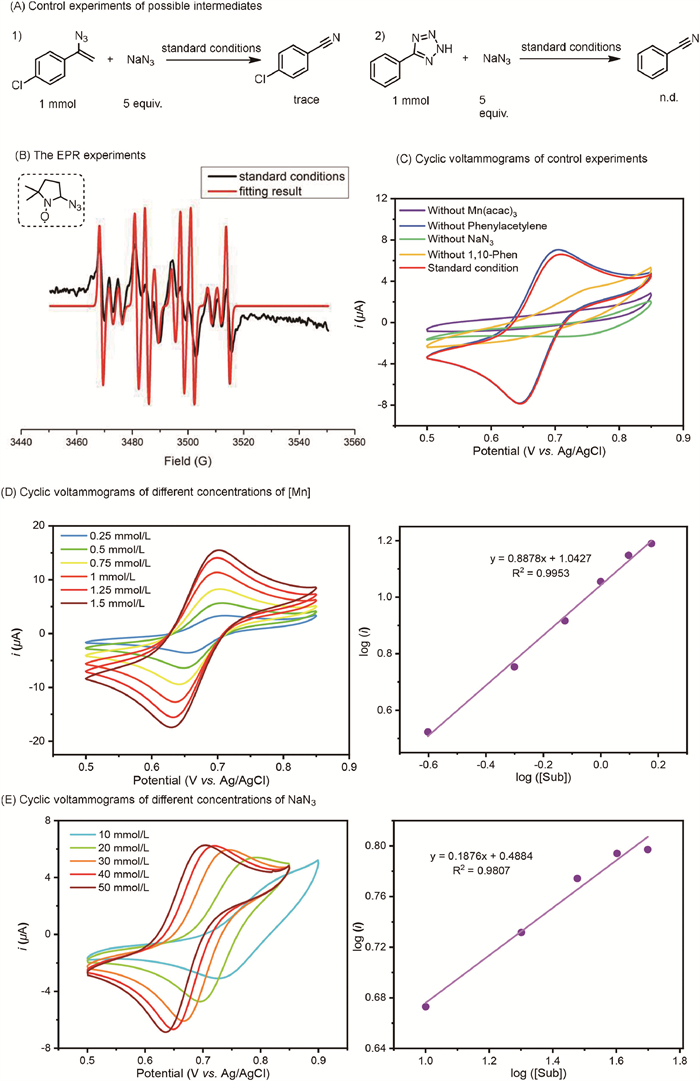
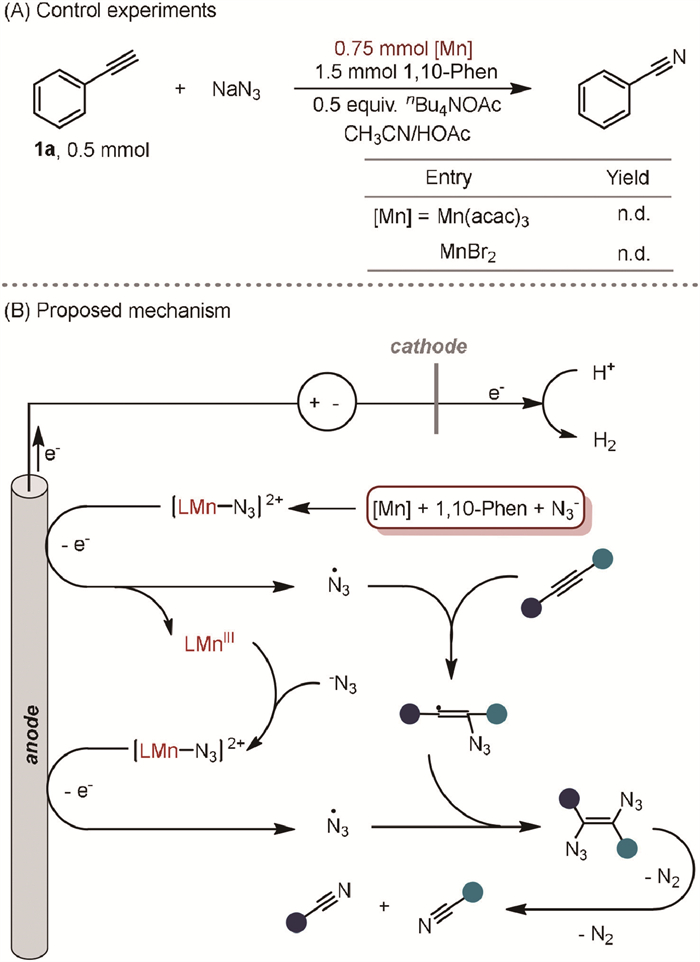
Subsequently, control experiments were conducted to gain deeper insights into potential intermediates in this transformation (Fig. 1A). When 1-(1-azidovinyl)-4-chlorobenzene (5a) and 5-phenyl-2H-tetrazole (5b) were used as substrates, the reactions did not proceed, indicating that vinyl azide and 2H-tetrazole were not intermediates in the reaction.

|
Download:
|
| Fig. 1. Mechanistic studies. (A) Control experiments of possible intermediates. (B) The EPR experiments. (C) Cyclic voltammograms experiments of control experiments. (D) Cyclic voltammograms experiments of different concentrations of Mn(acac)3. (E) Cyclic voltammograms experiments of different concentrations of NaN3. See Supporting information for further details. | |
{kind=link}
To further elucidate the mechanism, electron paramagnetic resonance (EPR) experiments were conducted to investigate radical intermediates. Under standard conditions, the azide radical (g = 2.0072, AN = 13.1 G, AH = 13.1 G, AN(β) = 3.4 G) was successfully captured using DMPO (5,5-dimethyl-1-pyrroline N-oxide) as the radical trapping reagent (Fig. 1B). In the absence of alkynes, the signal intensity of the azide radical significantly increased, suggesting a rapid reaction between the azide radicals and alkynes. Additionally, cyclic voltammetry (CV) experiments were conducted. The results revealed an oxidation peak around 0.7 V under standard conditions, consistently observed at different scan rates (Fig. S1 in Supporting information). It is reported that NaN3 can convert to azide radicals in the presence of HOAc, showing an oxidation peak around 1.5 V [52], which differs from the measured oxidation peaks. Consequently, control experiments were performed to verify the properties of this oxidation peak (Fig. 1C). These experiments showed that Mn(acac)3, NaN3, and 1,10-phenanthroline are essential for the reversible redox peaks, while phenylacetylene does not influence these peaks. Based on previously reported results [54] and our CV test results using [Mn(Ⅱ)] (Figs. S4 and S5 in Supporting information), the combination of Mn(acac)3, 1,10-phenanthroline, and NaN3 generates the redox pair L-Mn(Ⅲ)-N3 and L-Mn(Ⅱ)-N3 under CV measurement.
Further kinetic behavior using CV was measured at varying concentrations of Mn(acac)3 and NaN3 to investigate the mechanism. Increasing the concentration of the Mn catalyst enhanced the reaction activity (Fig. 1D). These findings suggest that NaN3 and 1,10-phenanthroline interact with Mn catalysts to facilitate the reaction. The reactivity positively correlates with the concentration of NaN3, whereas it negatively correlates with the concentration of 1,10-phenanthroline (Fig. 1E and Fig S3 in Supporting information). This phenomenon is likely due to competitive binding between NaN3 and 1,10-phenanthroline. Since 1,10-phenanthroline has a stronger coordinating ability with Mn atoms, its excess can poison the manganese catalysts, reducing reaction activity.
To verify whether Mn(acac)3 can be reduced by NaN3 to Mn(Ⅱ) and an N3 radical, experiments with stoichiometric amounts of manganese catalysts without electricity were performed (Scheme 4A). However, the reaction did not proceed when an excessive amount of either Mn(Ⅲ) or Mn(Ⅱ) manganese catalyst was added. These results suggest an essential interaction between the applied electric current and the formed Mn(Ⅱ)-N3 complex, enabling the reaction to proceed smoothly.

|
Download:
|
| Scheme 4. Control experiments and proposed mechanism. Reaction conditions: phenylacetylene (0.5 mmol), NaN3 (2.5 mmol), Mn(acac)3 (0.75 mmol), 1,10-Phen (1.5 mmol), and nBu4NOAc in CH3CN (4.25 mL)/HOAc (0.75 mL), stirring under nitrogen atmosphere at 25 ℃ for 11 h. | |
{kind=link}
Based on the experiments, a catalytic cycle for the electrochemical Mn-catalyzed cleavage of the alkyne triple bond (Scheme 4B). Initially, [Mn(Ⅱ)] coordinates with 1,10-phenanthroline, forming [LMn(Ⅱ)-N3] species in the presence of N3⁻. Anodic oxidation of [LMn(Ⅱ)-N3] then generates [LMn(Ⅲ)] and ·N3 radicals. These radicals add to the alkyne, producing an alkenyl radical. This alkenyl radical subsequently reacts with another ·N3 radical to form a 1,2-diazidovinyl compound, which undergoes rapid denitrification rearrangement, breaking the triple bond and yielding the final nitrile products. Meanwhile, at the cathode, protons are reduced to H₂.
In conclusion, we have developed a manganese-catalyzed electrochemical method that converts alkynes to nitriles under mild conditions. This approach applies to both terminal and a variety of internal alkynes, and it is compatible with many heterocyclic compounds. The ease of operation, broad functional group tolerance, and scalability of this method for nitrile generation via C≡C bond cleavage make it a valuable tool for molecular editing. We anticipate that this technique will be beneficial for chemical synthesis in academic research as well as in the pharmaceutical, material, and agrochemical industries.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementYuwei Liang: Methodology, Investigation, Conceptualization. Jianwei Huang: Methodology. Zhiqiang Zhang: Methodology. Qinghong Yang: Project administration, Methodology, Investigation, Conceptualization. Aiwen Lei: Project administration, Methodology, Investigation, Conceptualization. Hong Yi: Project administration, Methodology, Investigation, Conceptualization.
AcknowledgementsThis work was supported by the National Key R&D Program of China (Nos. 2022YFA1505100, 2021YFA1500104), National Natural Science Foundation of China (No. 22031008), Science Foundation of Wuhan (Nos. 2023020201020266, 2020010601012192), National nature science foundation of China (No. 22201222) and Jiangxi Normal University doctoral research initiation Fund project (Nos. 12017081, 12022796).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111166.
| [1] |
P.S. Baran, J. Am. Chem. Soc. 140 (2018) 4751-4755. DOI:10.1021/jacs.8b02266 |
| [2] |
W. Liu, B. Hong, J. Wang, X. Lei, Acc. Chem. Res. 53 (2020) 2569-2586. DOI:10.1021/acs.accounts.0c00531 |
| [3] |
A.G. Atanasov, S.B. Zotchev, V.M. Dirsch, C.T. Supuran, Nat. Rev. Drug. Discov. 20 (2021) 200-216. DOI:10.1038/s41573-020-00114-z |
| [4] |
S.H. Kennedy, B.D. Dherange, K.J. Berger, M.D. Levin, Nature 593 (2021) 223-227. DOI:10.1038/s41586-021-03448-9 |
| [5] |
Z. Fan, X. Chen, K. Tanaka, et al., Nature 610 (2022) 87-93. DOI:10.1038/s41586-022-05175-1 |
| [6] |
C. Hui, Z. Wang, S. Wang, C. Xu, Org. Chem. Front. 9 (2022) 1451-1457. DOI:10.1039/d2qo00043a |
| [7] |
P. Ji, C.C. Davies, F. Gao, et al., Nat. Commun. 13 (2022) 4565. DOI:10.1038/s41467-022-32201-7 |
| [8] |
J. Jurczyk, J. Woo, S.F. Kim, et al., Nat. Synth. 1 (2022) 352-364. DOI:10.1038/s44160-022-00052-1 |
| [9] |
B.A. Wright, A. Matviitsuk, M.J. Black, et al., J. Am. Chem. Soc. 145 (2023) 10960-10966. DOI:10.1021/jacs.3c02616 |
| [10] |
Z. Dong, Z. Ren, S.J. Thompson, Y. Xu, G. Dong, Chem. Rev. 117 (2017) 9333-9403. DOI:10.1021/acs.chemrev.6b00574 |
| [11] |
J. He, M. Wasa, K.S. Chan, Q. Shao, J.Q. Yu, Chem. Rev. 117 (2017) 8754-8786. DOI:10.1021/acs.chemrev.6b00622 |
| [12] |
Y. Park, Y. Kim, S. Chang, Chem. Rev. 117 (2017) 9247-9301. DOI:10.1021/acs.chemrev.6b00644 |
| [13] |
Y. Qin, L. Zhu, S. Luo, Chem. Rev. 117 (2017) 9433-9520. DOI:10.1021/acs.chemrev.6b00657 |
| [14] |
Y. Yang, J. Lan, J. You, Chem. Rev. 117 (2017) 8787-8863. DOI:10.1021/acs.chemrev.6b00567 |
| [15] |
H. Yi, G. Zhang, H. Wang, et al., Chem. Rev. 117 (2017) 9016-9085. DOI:10.1021/acs.chemrev.6b00620 |
| [16] |
P. Gandeepan, T. Müller, D. Zell, et al., Chem. Rev. 119 (2018) 2192-2452. |
| [17] |
S.K. Sinha, S. Guin, S. Maiti, et al., Chem. Rev. 122 (2021) 5682-5841. |
| [18] |
C.X. Ye, E. Meggers, Acc. Chem. Res. 56 (2023) 1128-1141. DOI:10.1021/acs.accounts.3c00081 |
| [19] |
S. Kim, J. Chung, B.M. Kim, Tetrahedron Lett. 52 (2011) 1363-1367. DOI:10.1016/j.tetlet.2011.01.065 |
| [20] |
Y. Deng, X.J. Wei, H. Wang, et al., Angew. Chem. Int. Ed. 56 (2017) 832-836. DOI:10.1002/anie.201607948 |
| [21] |
Z. Huang, R. Guan, M. Shanmugam, et al., J. Am. Chem. Soc. 143 (2021) 10005-10013. DOI:10.1021/jacs.1c05757 |
| [22] |
J. Li, J. Zhao, C. Ma, et al., ChemSusChem 14 (2021) 4985-4992. DOI:10.1002/cssc.202101504 |
| [23] |
Wang H., Jia R., Hong M., et al., Green Chem. 23 (2021) 6591-6597. DOI:10.1039/d1gc01931g |
| [24] |
A. Ruffoni, C. Hampton, M. Simonetti, D. Leonori, Nature 610 (2022) 81-86. DOI:10.1038/s41586-022-05211-0 |
| [25] |
F. Chen, T. Wang, N. Jiao, Chem. Rev. 114 (2014) 8613-8661. DOI:10.1021/cr400628s |
| [26] |
X. Zhu, J. Liu, W. Zhang, Nat. Chem. Biol. 11 (2015) 115-120. DOI:10.1038/nchembio.1718 |
| [27] |
F. Amblard, J.H. Cho, R.F. Schinazi, Chem. Rev. 109 (2009) 4207-4220. DOI:10.1021/cr9001462 |
| [28] |
K. Gilmore, I.V. Alabugin, Chem. Rev. 111 (2011) 6513-6556. DOI:10.1021/cr200164y |
| [29] |
R. Dorel, A.M. Echavarren, Chem. Rev. 115 (2015) 9028-9072. DOI:10.1021/cr500691k |
| [30] |
V.P. Boyarskiy, D.S. Ryabukhin, N.A. Bokach, A.V. Vasilyev, Chem. Rev. 116 (2016) 5894-5986. DOI:10.1021/acs.chemrev.5b00514 |
| [31] |
T. Shen, T. Wang, C. Qin, N. Jiao, Angew. Chem. Int. Ed. 52 (2013) 6677-6680. DOI:10.1002/anie.201300193 |
| [32] |
N. Okamoto, M. Ishikura, R. Yanada, Org. Lett. 15 (2013) 2571-2573. DOI:10.1021/ol401311h |
| [33] |
U. Dutta, D.W. Lupton, D. Maiti, Org. Lett. 18 (2016) 860-863. DOI:10.1021/acs.orglett.6b00147 |
| [34] |
Y. Lin, Q. Song, Eur. J. Org. Chem. 2016 (2016) 3056-3059. DOI:10.1002/ejoc.201600438 |
| [35] |
D.J. Constable, P.J. Dunn, J.D. Hayler, et al., Green Chem. 9 (2007) 411-420. DOI:10.1039/B703488C |
| [36] |
F. Fleming, Nat. Prod. Rep. 16 (1999) 597-606. DOI:10.1039/a804370a |
| [37] |
F.F. Fleming, L. Yao, P. Ravikumar, L. Funk, B.C. Shook, J. Med. Chem. 53 (2010) 7902-7917. DOI:10.1021/jm100762r |
| [38] |
X. Liu, L. Huang, Y. Ma, et al., Nat. Commun. 15 (2024) 7012. DOI:10.1038/s41467-024-51307-8 |
| [39] |
J. Qin, B. Han, X. Liu, et al., Sci. Adv. 8 (2022) eadd1267. DOI:10.1126/sciadv.add1267 |
| [40] |
C. Xian, J. He, Y. He, et al., J. Am. Chem. Soc. 144 (2022) 23321-23331. DOI:10.1021/jacs.2c07061 |
| [41] |
J.B. Sperry, D.L. Wright, Chem. Soc. Rev. 35 (2006) 605-621. DOI:10.1039/b512308a |
| [42] |
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230-13319. DOI:10.1021/acs.chemrev.7b00397 |
| [43] |
Q.L. Yang, P. Fang, T.S. Mei, Chin. J. Chem. 36 (2018) 338-352. DOI:10.1002/cjoc.201700740 |
| [44] |
Q. Jing, K.D. Moeller, Acc. Chem. Res. 53 (2019) 135-143. |
| [45] |
N. Chen, H.C. Xu, Chem. Rec. 21 (2021) 2306-2319. DOI:10.1002/tcr.202100048 |
| [46] |
L.F. Novaes, J. Liu, Y. Shen, et al., Chem. Soc. Rev. 50 (2021) 7941-8002. DOI:10.1039/d1cs00223f |
| [47] |
Z. Yang, W. Shi, H. Alhumade, H. Yi, A. Lei, Nat. Synth. 2 (2023) 217-230. DOI:10.1038/s44160-022-00221-2 |
| [48] |
P. Wang, S. Tang, P. Huang, A. Lei, Angew. Chem. 129 (2017) 3055-3059. DOI:10.1002/ange.201700012 |
| [49] |
L. Jie, B. Guo, J. Song, H. Xu, J. Am. Chem. Soc. 144 (2022) 2343-2350. DOI:10.1021/jacs.1c12762 |
| [50] |
X. Liu, D. Yang, Z. Liu, et al., J. Am. Chem. Soc. 145 (2023) 3175-3186. DOI:10.1021/jacs.2c12902 |
| [51] |
N. Fu, G.S. Sauer, A. Saha, A. Loo, S. Lin, Science 357 (2017) 575-579. DOI:10.1126/science.aan6206 |
| [52] |
L. Niu, C. Jiang, Y. Liang, et al., J. Am. Chem. Soc. 142 (2020) 17693-17702. DOI:10.1021/jacs.0c08437 |
| [53] |
T.H. Meyer, R.C. Samanta, A. Del Vecchio, L. Ackermann, Chem. Sci. 12 (2021) 2890-2897. DOI:10.1039/d0sc05924b |
| [54] |
L.F. Novaes, Y. Wang, J. Liu, et al., ACS Catal. 12 (2022) 14106-14112. DOI:10.1021/acscatal.2c05186 |
| [55] |
Y. Wang, L. Li, N. Fu, ACS Catal. 12 (2022) 10661-10667. DOI:10.1021/acscatal.2c02934 |
| [56] |
Y. Weng, X. Xu, H. Chen, Y. Zhang, X. Zhuo, Angew. Chem. 134 (2022) e202206308. DOI:10.1002/ange.202206308 |
| [57] |
M. Cao, H. Wang, Y. Ma, C.H. Tung, L. Liu, J. Am. Chem. Soc. 144 (2022) 15383-15390. DOI:10.1021/jacs.2c07089 |
| [58] |
X. Huang, T.M. Bergsten, J.T. Groves, J. Am. Chem. Soc. 137 (2015) 5300-5303. DOI:10.1021/jacs.5b01983 |
| [59] |
L.M. Jin, P. Xu, J. Xie, X.P. Zhang, J. Am. Chem. Soc. 142 (2020) 20828-20836. DOI:10.1021/jacs.0c10415 |
| [60] |
X. Kong, Y. Liu, L. Lin, Q. Chen, B. Xu, Green Chem. 21 (2019) 3796–3801.
|
| [61] |
S. Li, H.W. Du, P.W. Davies, W. Shu, CCS Chem. 6 (2024) 1060-1070. DOI:10.31635/ccschem.023.202302999 |
| [62] |
O.M. Mulina, N.V. Zhironkina, S.A. Paveliev, D.V. Demchuk, A.O. Terent'ev, Org. Lett. 22 (2020) 1818-1824. DOI:10.1021/acs.orglett.0c00139 |
| [63] |
Y. Ning, X.F. Zhao, Y.B. Wu, X. Bi, Org. Lett. 19 (2017) 6240-6243. DOI:10.1021/acs.orglett.7b03204 |