 2026, Vol. 37
2026, Vol. 37 


b ZJU-Hangzhou Global Scientific and Technological Innovation Center, Zhejiang University, Hangzhou 311200, China;
c Homogeneous, Supramolecular and Bio-Inspired Catalysis, Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Amsterdam 1098XH, The Netherlands
The advent of macrocyclic molecules marks an outstanding milestone in the history of supramolecular chemistry [1-3]. This is because these hosts can take advantage of their inherent cavities to encapsulate guest molecules, altering their properties without disturbing their own covalent structures [4-8]. As a consequence, a variety of tasks could be accomplished in a supramolecular manner, such as stabilizing guests or reaction intermediates [9-15], molecular sensing [16-19], separating guests [20-28]. Thus the design and synthesis of macrocyclic hosts have become one of the major focuses in the field of supramolecular chemistry. The traditionally approaches rely on irreversible organic reactions, such as SN2 reactions in the synthesis of crown ethers [29]. However, "errors" often occur in covalent bond connections, leading to the formation of oligomeric and polymeric byproducts, making irreversible reactions not suitable for synthesizing more complex target molecules during which multiple chemical bonds (number of bonds > 2) are formed. Using reversible reactions including the formation of coordinative [30-32] and dynamic covalent bonds [33-42] allows error checking. Hence, the target molecules representing the thermodynamic minima can often be self-assembled in high or close to quantitative yield. The drawback of self-assembly, however, is also remarkable. That is, the molecules containing dynamic bonds often or always undergo degradation via bond exchange or cleavage [43]. In addition, the labile nature of dynamic molecules also jeopardizes the opportunity of post-functionalization of these self-assembled molecules.
Here, by taking advantage of the dynamic nature of Friedel–Crafts (FC) reaction [44-49] between furan and olefin in the presence of Lewis acid catalyst, a triangular prismatic molecular cage was obtained in a one-pot manner in a modest (i.e., 25%) but satisfactory yield, when considering the fact that six C‒C bonds were formed during ring closure step. The cage framework contains two trisfuran platforms bridged by three phenanthrene pillars each bearing two electron-donating methoxy units. Consequently, the cage can accommodate planar π-electron deficient guests by utilizing two of its three phenanthrene pillars. Notably, in the absence of any Lewis acid catalysts in FC reaction, this cage is kinetically inert. This advantage affords an opportunity for post-functionalization without degradation of cage framework. After removing all methoxy units and the following oxidation process, all the methoxy units in the cage framework were transferred into carbonyl units that are rather electron deficient, which altered the guest recognition preference of the cage. As a consequence, the cage can recognize π-electron rich guests such as pyrene. In both forms, the cage can utilize the oxygen atoms in either methoxy or carbonyl units as Lewis basic binding sites to grab alkali cations. Upon the recognition of alkali cationic guests, a dramatic conformational switch occurred in the cage framework. This binding process yields a well-established concept reminiscence of the enzymes functional process which undergoing induced-fit behaviour when accommodating target substrates.
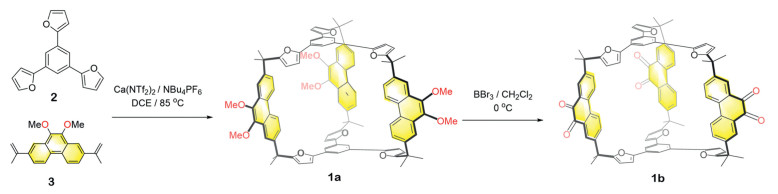
The synthesis of cage 1a was carried out by performing FC reaction on a 2:3 mixture of a trisfuran precursor 2 and a phenanthrene derivative 3 in 1,2-dichloroethane (DCE), using Ca(NTf2)2 as a Lewis acid catalyst. NBu4PF6 was added into the reaction mixture to enhance the solubility or reactivity of Ca2+ [50]. After heating the mixture for 3 h, the crude product was purified via column chromatography, yielding the compound 1a as a pure product with a 25% yield (Fig. 1). Cage 1a exhibits good solubility in many commonly-used solvents such as chloroform, dichloromethane (DCM), toluene and dimethylformamide (DMF). The structure of cage 1a was fully characterized via nuclear magnetic resonance spectroscopy (NMR) and high-resolution mass spectrometry (HRMS) (Figs. S2-S8 in Supporting information).

|
Download:
|
| Fig. 1. Synthesis of 1a by performing Friedel–Crafts reaction to a 3:2 mixture of 2 and 3, following by the post-functionalization to obtain 1b. | |
{kind=link}
Upon removal of calcium catalyst during purification, the cage framework of 1a is demonstrated rather stable and kinetically inert. For example, no observable degradation occurred after heating 1a in either solid form or in a toluene solution for at least 3 days. This stability encouraged us to perform post-functionalization on 1a. The addition of boron tribromide (BBr3) into a DCM solution of 1a deprotected all the methoxy (MeO) groups and converted them into hydroxyl (OH) groups. Hydroxyl (OH) groups were subsequently oxidized within a few hours into carbonyl units, yielding cage 1b, which bears six carbonyl units. Cage 1b was isolated in a 91% yield after purification via precipitation after the addition of methanol (Fig. 1).
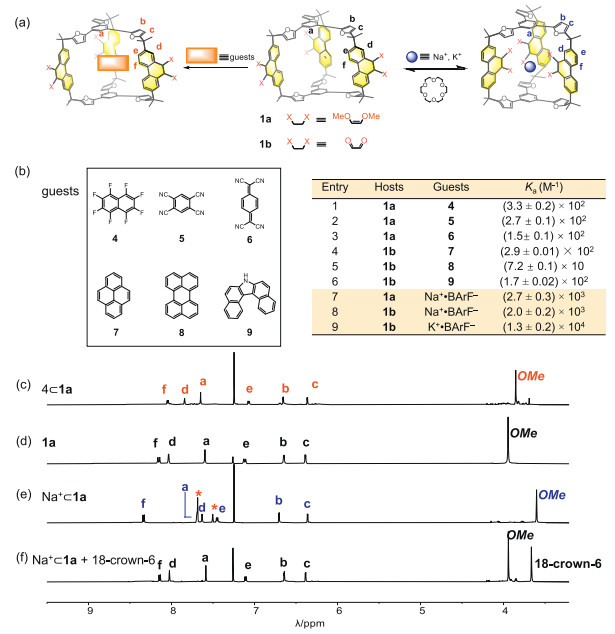
The abilities of both cage 1a and 1b to encapsulate guests were then investigated. Cage 1a, containing electron donating methoxy units, encouraged us to explore the recognition of electron deficient guests, by taking advantage of either π-electron donor-acceptor interactions or dipole-dipole forces. Upon addition of octafluoronaphthalene (4) into a CDCl3 solution of 1a, the resonances corresponding to the protons d and f in the phenanthrene pillars underwent upfield shifts (-0.20 ppm and -0.10 ppm respectively), while other protons exhibited minimal shifts (Fig. 2c). This observation indicates that guest 4 is recognized within the cavity of the cage 1a, by interacting with the phenanthrene pillars (Figs. S22 and S23 in Supporting information). In comparison, cage 1a utilizes the trisfuran platforms to recognize both 1,2,4,5-tetracyanobenzene (5) and tetracyanoquinodimethane (6), as indicated by the observation that addition of these two guests led to modest upfield shifts of the phenyl units in the central part of the trisfuran platforms (Figs. S24 and S26 in Supporting information). The addition of electron rich guests, including pyrene (7), 7H-dibenzo[c,g]carbazole (8), and perylene (9), into the solution of 1a, led to no observable shifts, indicating that cage 1a bearing methoxy groups has an electron rich cavity, incapable of recognizing electron rich substrates due to the absence of supramolecular driving forces such as dipole-dipole or donor-acceptor interactions (Fig. S28 in Supporting information).

|
Download:
|
| Fig. 2. (a) Schematic representation of two typical strategies of recognition. Left: recognition of planar guests by platforms. Right: recognition of alkali cations by basic binding sites. (b) Left: a series of planar guests 4-9. Right: association constants (Ka) of 1a and 1b towards guests. Partial 1H NMR spectra (500 MHz, CDCl3, 298 K) of (c) 4⊂1a, (d) 1a, (e) Na+⊂1a and (f) addition 1 equiv. 18-crown-6 to Na+⊂1a (* represents the peak of BArF− in solution). | |
{kind=link}
In the case of cage 1b, as a comparison, it binds well with the electron rich guests 7, 8 and 9, by taking advantage of the electron deficiency of the pillars bearing carbonyl units. This is confirmed by the observation that upon the addition of each of these guests, the resonances corresponding to the protons e and f in the phenanthrene pillars underwent upfield shifts (Figs. S30, S32 and S34 in Supporting information). Conversely, the addition of the electron-deficient guests 4, 5 and 6 into the solution of 1b, led no observable shifts in the 1H NMR spectra (Fig. S35 in Supporting information), confirming that 1b can only recognize electron-rich guests. This implies that switching the electronic properties of the phenanthrene pillars altered the guest recognition preference of the cage.
Each phenanthrene pillar, contains two oxygen atoms in the form of either methoxy or carbonyl in cage 1a and 1b respectively. These oxygen atoms are typical Lewis basic binding sites. This encourages us to investigate the potential of using the cage in both forms to accommodate alkali cations, which are typical Lewis acidic guests. The addition of sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (Na+·BArF‒) into a solution of 1a in CDCl3 led to remarkable shifts of the protons in the phenanthrene pillars. The resonances corresponding to e and f shifted downfield (+0.26 ppm and +0.19 ppm respectively), while those corresponding to proton d and proton of methyl apparently shifted upfield (-0.40 ppm and -0.64 ppm respectively) (Fig. 2e). The differences result from the guest induced conformational change of the cage 1a. In the absence of cationic guest each the phenanthrene pillar undergoes a pivoting motion within the cage framework. On the one hand, upon recognition of Na+, all the methoxy units reorient inward, to form dipole-cation interactions in the form of O‒Na+. Protons d located next to methoxy units are thus shaded inside the cage cavity, experiencing a shielded magnetic environment. On the other hand, protons e and f are forced to point outward, thus, experiencing a de-shielded magnetic environment (Fig. S41 in Supporting information). By performing 1H NMR titration experiments, the binding constant of the complex Na+⊂1a was measured to be (2.7 ± 0.3) × 103 L/mol. The addition of either smaller cationic guest such as Li+, or larger counterparts like K+ and Cs+, resulted in minimal shifts of the corresponding resonances in the 1H NMR spectra (Fig. S40 in Supporting information), indicating these cations do not possess complementary sizes to fit within the cage cavity. The cationic guest in the complex Na+⊂1a can be removed by adding 18-crown-6, after which the 1H NMR spectrum was restored (Fig. 2f, Figs. S56 and S57 in Supporting information), indicating the pivoting motion of the phenanthrene units was reactivated upon removing the Na+ guest. In the case of cage 1b, both Na+ and K+ were recognized within the cage cavity, as inferred from the corresponding 1H NMR spectra. The binding constants of Na+⊂1b and K+⊂1b were measured to be (2.0 ± 0.2) × 103 and (1.3 ± 0.2) × 104 L/mol, respectively. Apparently, the removal of the methyl units from 1a increases the effective volumes of the intrinsic cavity of the cage 1b, making the size of its cavity complementary to K+. The addition of crown ether led to similar observations that the conformation of the free cage 1b was recovered (Figs. S60 and S61 in Supporting information).
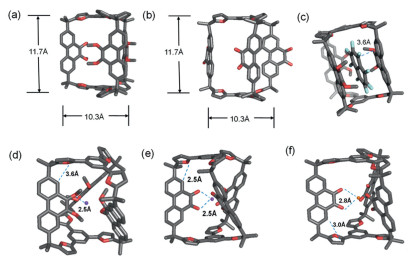
To further elucidate the structures of both the cage and its complexes, diffraction grade single crystals of 1a were obtained by the slow vapor diffusion of methanol into its solution in DCM (CCDC: 2414758). The single crystals of 1b and 4⊂1a were obtained by vapor diffusion of methyl t-butyl ether into their solutions in chloroform (CCDC: 2414759 and 2414760). These solid-state structures provided unambiguous evidence of their configurations. As expected, both 1a and 1b exhibit triangular prism-shaped framework in solid-state. In the case of 1a, all the methoxy units point inward (Fig. 3a and Figs. S15-S18 in Supporting information), while in 1b, all the carbonyl units point outward (Fig. 3b and Figs. S19-S21 in Supporting information). In the framework of 4⊂1a, guest 4 inserts into the gap between two of the three phenanthrene pillars, forming a sandwich-shaped structure. The distance between guest 4 and each of the two adjacent phenanthrene pillars was measured to be approximately 3.6 Å (Fig. 3c), a typical π-π interaction distance which is well consistent with 1H NMR spectroscopic results, indicating the cage 1a utilizes its pillars to recognize guest 4.

|
Download:
|
| Fig. 3. Single crystal X-ray diffraction structures of the cage (a) 1a, (b) 1b. (c) Single crystal X-ray diffraction structures of the host-guest complex 4⊂1a. Optimized structure of (d) Na+⊂1a, (e) Na+⊂1b, and (f) K+⊂1b obtained by DFT calculations. Color code: C gray, O red, H white, Na purple, K orange. disordered counterions, solvent molecules have been omitted for clarity. | |
{kind=link}
Attempts to obtain the diffraction grade single crystals of complexes containing alkali cations were turned to be unsuccessful. Therefore, density functional theory (DFT) calculations were performed to elucidate the structures of these complexes. As expected, in the optimized structures of Na+⊂1a, Na+⊂1b and K+⊂1b, all the Lewis basic binding sites, namely the oxygen atoms in either methoxy or carbonyl units, point inward to coordinate with the cationic alkali guests (Figs. 3d-f). The MeO–Na+ distance was determined to be approximately 2.5 Å, indicating chelated dipole–cation interactions in 1a. it was observed that the coordination of Na+, which drives the cation from an outward to an inward position, is a highly favorable process. The Gibbs free energy change (ΔG) was calculated to be −77.2 kcal/mol using DFT (Fig. S63 in Supporting information). Similarly, upon the addition of Na+ or K+ to 1b, the calculated ΔG values were −56.4 kcal/mol and −48.3 kcal/mol, respectively, indicating spontaneous recognition of Na+⊂1b and K+⊂1b (Figs. S65 and S67 in Supporting information).
In summary, by leveraging the Friedel–Crafts (FC) reaction, a triangular prismatic cage was synthesized with a modest yield. Upon removal of the Lewis acidic catalyst during purification, the cage exhibited significant kinetic inertness, allowing for post-functionalization without degradation of the cage framework. The methoxy groups were thus transformed into carbonyl units, by removing the methyl during which oxidation occurred.
The cage, in both forms, demonstrates multiple guest binding modes with conformational adaptive capability. For example, both pre- and post-functionalized cages can utilize their oxygen atoms in either methoxy or carbonyl units to recognize Lewis acidic guests, namely Na+ and K+. A remarkable conformation switching was observed upon guest accommodation, whereby the cage can rotate its phenanthrene pillars to direct its oxygen binding sites to point inwards, forming cation-dipole interactions with the alkali guests. Post-functionalization also alters the guest accommodation preference of the cage in recognition of π-electron rich and deficient guests. Specifically, the cages bearing methoxy and carbonyl units can preferentially accommodate π-electron deficient and rich guests respectively.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementYongwei Qian: Visualization, Software, Methodology, Investigation, Formal analysis, Data curation. Shengyang Huang: Validation, Software, Methodology, Investigation, Data curation. Yaoyi Su: Validation, Software. Songna Zhang: Writing – original draft. Yang Liu: Project administration, Methodology. Ming Liu: Supervision. Bin Sun: Writing – review & editing, Conceptualization. Hao Li: Writing – review & editing, Writing – original draft, Conceptualization.
AcknowledgmentsThe research at Zhejiang University was supported by the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (No. SN-ZJU-SIAS-006). H. Li also want to thank the support from the Leading Innovation Team grant from Department of Science and Technology of Zhejiang Province (No. 2022R01005), the Natural Science Foundation of Zhejiang Province (No. LZ24B020002) and the National Natural Science Foundation of China (No. 22471240). We thank Prof. Qiaohong He, Dr. Jiyong Liu, Dr. Yaqin Liu, Dr. Lina Gao and Dr. Yifan Zhao from the Chemistry Instrumentation Center Zhejiang University for the technical support.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111354.
| [1] |
J.M. Lehn, Science 260 (1993) 1762-1763. DOI:10.1126/science.8511582 |
| [2] |
D.J. Cram, T.K. Roger, C. Helgeson, G.M. Lein, J. Am. Chem. Soc. 101 (1979) 6752-6754. DOI:10.1021/ja00516a048 |
| [3] |
D.J. Cram, Kaneda T, R.C. Helgeson, G.M. Lein, J. Am. Chem. Soc. 89 (1967) 2495-2496. DOI:10.1021/ja00986a052 |
| [4] |
Y.H. Ko, H. Kim, Y. Kim, K. Kim, Angew. Chem. Int. Ed. 47 (2008) 4106-4109. DOI:10.1002/anie.200800581 |
| [5] |
K. Ariga, H. Ito, J.P. Hill, Chem. Soc. Rev. 41 (2012) 5800-5835. DOI:10.1039/c2cs35162e |
| [6] |
J. Rebek, J.Am Jr, Chem. Soc. 136 (2014) 5264-5266. DOI:10.1021/ja501685z |
| [7] |
F.U. Rahman, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 17648-17653. DOI:10.1073/pnas.1909154116 |
| [8] |
E.G. Percástegui, T.K. Ronson, J.R. Nitschke, Chem. Rev. 120 (2020) 13480-13544. DOI:10.1021/acs.chemrev.0c00672 |
| [9] |
M. Yamanaka, A. Shivanyuk, J. Rebek. Jr, J. Am. Chem. Soc. 126 (2004) 2939-2943. DOI:10.1021/ja037035u |
| [10] |
C.M. Hong, R.G. Bergman, K.N. Raymond, F.D. Toste, Acc. Chem. Res. 51 (2018) 2447-2455. DOI:10.1021/acs.accounts.8b00328 |
| [11] |
Q. Zhang, L. Catti, Tiefenbacher, Acc. Chem. Res. 51 (2018) 2107-2114. DOI:10.1021/acs.accounts.8b00320 |
| [12] |
J. Rebek. Jr, Y. Yu, Chem. Commun. 55 (2019) 3573-3577. DOI:10.1039/C9CC01611B |
| [13] |
T.A. Young, V.M. Centelles, J.Z Wang, F. Duarte, J. Am. Chem. Soc. 142 (2020) 1300-1310. DOI:10.1021/jacs.9b10302 |
| [14] |
J.M. Yang, Y. Yu, J. Rebek. Jr, J. Am. Chem. Soc. 143 (2021) 2190-2193. DOI:10.1021/jacs.0c12302 |
| [15] |
P.S. Mukherjee, Angew. Chem. Int. Ed. 62 (2023) e202305338. DOI:10.1002/anie.202305338 |
| [16] |
W.M Xuan, M.N. Zhang, Y. Liu, Z. Chen, Y. Cui, J. Am. Chem. Soc. 134 (2012) 6904-6907. DOI:10.1021/ja212132r |
| [17] |
N. Ahmad, H.A. Younus, A.H. Chughtai, Chem. Soc. Rev. 44 (2015) 9-25. DOI:10.1039/C4CS00222A |
| [18] |
C. Lu, X. Yan, X. Li, F. Huang, S. Yin, J. Am. Chem. Soc. 139 (2017) 5067-5074. DOI:10.1021/jacs.6b12536 |
| [19] |
Z. Zhang, X. Wang, J. Am. Chem. Soc. 142 (2020) 2592-2600. DOI:10.1021/jacs.9b12689 |
| [20] |
T. Tozawa, S.I. Swamy, S. Jiang, A. Steiner, A.I. Cooper, Nat. Mater. 8 (2009) 973-978. DOI:10.1038/nmat2545 |
| [21] |
K.Z. Su, W.J. Wang, S.F. Du, C.Q. Ji, D.Q. Yuan, J. Am. Chem. Soc. 142 (2020) 18060-18072. DOI:10.1021/jacs.0c07367 |
| [22] |
G. Wu, H. Li, J. Am. Chem. Soc. 140 (2018) 5955-5961. DOI:10.1021/jacs.8b01651 |
| [23] |
Y. Zhang, H. Li, CCS Chem. 7 (2025) 50-58. DOI:10.31635/ccschem.024.202404345 |
| [24] |
Y.J. Zhou, K.C. Jie, R. Zhao, E. Li, F.H. Huang, J. Am. Chem. Soc. 142 (2020) 6957-6961. DOI:10.1021/jacs.0c02684 |
| [25] |
J. Zhou, G.C Yu, Q. Li, M.B Wang, F.H. Huang, J. Am. Chem. Soc. 142 (2020) 2228-2232. DOI:10.1021/jacs.9b13548 |
| [26] |
Y.T. Wu, J. Zhou, E. Li, et al., J. Am. Chem. Soc. 142 (2020) 19722-19730. DOI:10.1021/jacs.0c09757 |
| [27] |
Y. Liu, F.H. Huang, J. Am. Chem. Soc. 143 (2021) 3081-3085. DOI:10.1021/jacs.1c01204 |
| [28] |
M. Wang, F.H. Huang, Mater. Today Chem. 24 (2022) 2-8. |
| [29] |
C.J. Pedersen, J. Am. Chem. Soc. 89 (1967) 7017. DOI:10.1021/ja01002a035 |
| [30] |
M. Fujita, Nature 378 (1996) 469-471. |
| [31] |
M. Fujita, O. Katsuyuki, Nature 400 (1999) 53-55. |
| [32] |
M. Fujita, Nature 398 (1999) 794-796. DOI:10.1038/19734 |
| [33] |
D.J. Cram, J. Am. Chem. Soc. 113 (1991) 2754-2755. DOI:10.1021/ja00007a060 |
| [34] |
M. Mastalerz, Chem. Commun. (2008) 4756-4758. DOI:10.1039/b808990f |
| [35] |
Y. Liu, G. Li, X. Liu, R. Warmuth, Angew. Chem. Int. Ed. 45 (2006) 901-904. DOI:10.1002/anie.200504049 |
| [36] |
Y. Chen, H. Tang, H.L. Chen, H. Li, Acc. Chem. Res. 56 (2023) 2838-2850. DOI:10.1021/acs.accounts.3c00475 |
| [37] |
S.J. Rowan, P.A. Brady, M. Sanders, Angew. Chem. Int. Ed. 35 (1996) 2143-2145. DOI:10.1002/anie.199621431 |
| [38] |
S. Otto, M. Sanders, Science 297 (2002) 590-593. DOI:10.1126/science.1072361 |
| [39] |
M. Sanders, Angew. Chem. Int. Ed. 108 (2006) 2283-2285. |
| [40] |
L.Y. Wang, V. BÖhmer, Science 304 (2004) 1312-1314. DOI:10.1126/science.1096688 |
| [41] |
P.H. Kirchner, F. Beuerle, J. Am. Chem. Soc. 146 (2024) 5305-5315. DOI:10.1021/jacs.3c12002 |
| [42] |
G. Zhang, O.M.M.A Mastalerz, Angew. Chem. Int. Ed. 53 (2014) 1516-1520. DOI:10.1002/anie.201308924 |
| [43] |
M.S. Avinash, S. Bhat, S.M. Elbert, et al., Angew. Chem. Int. Ed. 58 (2019) 8819-8823. DOI:10.1002/anie.201903631 |
| [44] |
K. Xu, Z.Y. Zhang, C.G. Yu, et al., Angew. Chem. Int. Ed. 59 (2020) 7214-7218. DOI:10.1002/anie.202000909 |
| [45] |
K.D. Xu, B. Li, S.B. Yao, et al., Angew. Chem. Int. Ed. 61 (2022) e202203016. DOI:10.1002/anie.202203016 |
| [46] |
X.N. Han, Y. Han, C.F. Chen, Chem. Soc. Rev. 52 (2023) 3265-3298. DOI:10.1039/d3cs00002h |
| [47] |
X. Zhao, H.Y. Cui, L.L. Guo, et al., Angew. Chem. Int. Ed. 63 (2024) e202411613. DOI:10.1002/anie.202411613 |
| [48] |
D.S. Zhu, F. Shuai, L. Tong, H. Li, Chem. Commun. 57 (2021) 4440-4443. DOI:10.1039/d1cc00498k |
| [49] |
D. Zhu, B. Sun, L. Tong, H. Li, Org. Lett. 24 (2022) 8980-8985. DOI:10.1021/acs.orglett.2c03560 |
| [50] |
C. Denis, M.A.J. Dubois, A.S. Voisin-Chiret, et al., Org. Lett. 21 (2019) 300-304. DOI:10.1021/acs.orglett.8b03745 |