 2025, Vol. 36
2025, Vol. 36 


Isatin, known for its alternative name indoline-2,3–dione, along with its derivatives, occupies a prestigious position in the realm of medicinal chemistry and organic synthesis [1-9]. A profound exploration into the reactivity of isatins, possessing an indole structure adorned with a ketone and a γ-lactam entity, has revealed numerous captivating facets of organic transformations [10,11]. The most enchanting application of isatins in organic chemistry stems from the remarkably reactive C-3 carbonyl group, which serves as a prochiral center [12-22]. The corresponding arylated derivatives of isatins, specifically enantiomerically pure 3-hydroxyoxindoles, emerge as abundant and indispensable constituents within a plethora of biologically active compounds and drugs (Fig. 1a) [23]. For instance, coprisidins B, extracted from a gut-associated Streptomyces sp. in the dung beetle Copris tripartitus, demonstrate activity in inducing NAD(P)H:quinone oxidoreductase 1 [24]. Another noteworthy example is SM-130686, a remarkably potent and orally active nonpeptidic growth hormone secretagogue [25]. Additionally, 3-hydroxyoxindoles assume crucial roles as fundamental intermediates in a variety of total syntheses, encompassing a remarkable molucule such as diazonamide A [26-29]. Hence, facile strategies for the asymmetric construction of the carbon center in 3-hydroxyoxindoles hold substantial significance.

|
Download:
|
| Fig. 1. Metal-catalyzed asymmetric synthesis of 3-hydroxyoxindoles through aryl addition. | |
The application of aromatic organometallic agents in the asymmetric addition to the C3 carbonyl group of isatins, facilitated by metal catalysts, is considered to be an exceptionally advantageous strategy for obtaining chiral quaternary carbon centers of 3-hydroxyoxindoles. The pioneering work carried out by Hayashi and coworkers in 2006 first introduced the rhodium-catalyzed asymmetric addition of arylboronic acids to isatins (Fig. 1b) [30]. However, the reaction exhibited low enantioselectivity when working with unsubstituted isatins at the 5-position [31]. Therefore, numerous investigations have been conducted in the field of asymmetric catalysis employing arylboronic acids and esters in order to overcome this limitation [32-42]. The utilization of alternative organometallic compounds such as arylsilanes has also demonstrated remarkable effectiveness in the synthesis of chiral 3-hydroxyoxindoles through aryl addition into isatins, facilitated by the presence of a copper catalyst [43]. Despite of these advances, heteroaryl organometallic compounds, whether in the form of boron or silane reagents, continues to present a formidable obstacle in these reactions [44]. Considering factors such as ease of use and cost, there is an increasing tendency towards approaches that avoid the utilization of organometallic compounds and noble metal catalysts, all the while maximizing compatibility with the functional groups found in the reactants involved in the coupling process.
The venerable Nozaki-Hiyama-Kishi (NHK) reaction, first discovered in 1977 [45] and formalized in 1986 [46], is one of the most useful and reliable C—C bond-forming reactions in organic synthesis (Fig. 1c). It generally involves the cross-coupling of an alkenyl halide with an aldehyde through the use of stoichiometric Cr and catalytic Ni to afford an allylic alcohol product. Substantial advancements have been achieved in the realm of catalytic asymmetric carbonyl addition reactions under mild reductive conditions, opening the path for the direct use of aromatic (pseudo)halides as substitutes for preformed organometallic reagents [47-57]. However, practical control of enantioselectivity in ketone addition has not been reported [58-68]. Herein, we present the groundbreaking catalytic asymmetric NHK type reactions of isatins with aromatic halides (Fig. 1d). Upon exposure to a cobalt catalyst [69-83] adorned with a commercially available chiral biphosphine ligand, isatins exhibit the capacity to undergo reductive 1,2-addition with aromatic iodides. By substituting conventional metal dust with bis(neopentyl glycolato)diboron (B2nep2) as a reductant, remarkable progress has been made in both yield enhancement and the preservation of enantioselectivity at elevated levels. In a remarkable showcase that sets it apart from prior methodologies, this chemistry has achieved an exceptional level of precision in governing chemoselectivity, favoring the reaction of aromatic C–I bonds instead of C–B bonds. Moreover, a diverse array of heteroaryl haildes proved to be compatible in this instance.
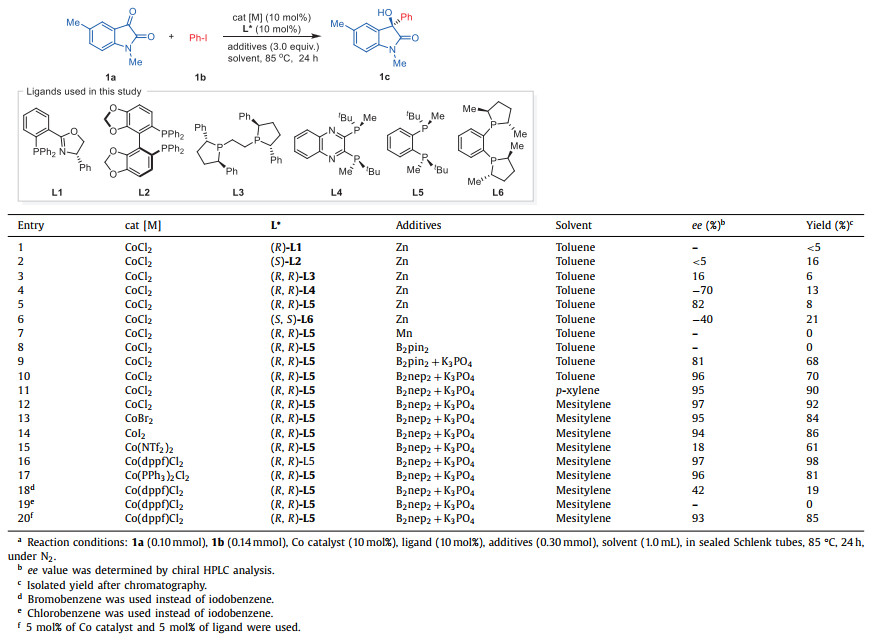
Our inquiries began with the reaction of isatin 1a and iodobenzene (1b) employing a catalytic amount of CoCl2 (10 mol%) and a stoichiometric quantity of zinc powder as the reducing agent in toluene at 85 ℃ (Table 1). The formation of the addition product 1c was only detectable in minute quantities when employing the pyridine-oxazoline ligand (R)-L1 (entry 1). Subsequent screening of various chiral ligands revealed that the utilization of (S)-SEGPHOS (L2) yielded the desired product 1c with a yield of 16%, but almost racemic (entry 2). Further exploration of bisphosphine ligands under optimal conditions confirmed that (R,R)-Ph-BPE (L3) provided a level of 16% ee (entry 3). It was with great satisfaction that we observed how the implementation of (R,R)-QuinoxP (L4) markedly enhanced the enantioselectivity, affording 1c with an 70% ee, despite the yield still being low (entry 4). The treatment of (R,R)-BenzP (L5) produced product 1c with exceptional enantioselectivity (82% ee), though the yield remained suboptimal (entry 5). Under the established reaction conditions, the use of (S,S)-DUPHOS (L6) merely produced modest levels of enantioselectivity (entry 6). When employing L5 as the optimical ligand, alternate reducing agents such as manganese dust proved entirely ineffective in this reaction (entry 7). Although the use of bis(pinacolato)diboron (B2pin2) did not yield the desired product (entry 8), the addition of K3PO4 to the system greatly enhanced the formation of 1c, resulting in a 68% yield and 81% ee (entry 9). It was discovered that treating bis(neopentyl glycolato)diboron (B2nep2) led to a favorable yield (90%), as well as an improved enantioselectivity of 96% (entry 10). To examine the influence of solvents, we also tested various options. P-xylene proved to be more effective, producing 1c with a 90% yield and maintaining enantioselectivity (entry 11). When mesitylene was utilized as the solvent, the reaction exhibited even better results (92% yield, 97% ee, entry 12). Alternative cobalt sources, such as CoBr2 and CoI2, also yielded 1c in excellent enantioselectivities, albeit with slightly lower yields (84%−86%, entries 13 and 14). Nonetheless, the utilization of Co(NTf2)2 resulted in remarkably diminished reactivity during the reaction (entry 15). Surprisingly, the complex of 1,1′-bis(diphenylphosphino)ferrocene, Co(dppf)Cl2, displayed remarkable reactivity, producing 1c with a 98% yield and 97% ee (entry 16). Another complex, Co(PPh3)2Cl2, also maintained excellent enantioselectivity (96% ee), albeit with a lower yield (81%, entry 17). To investigate the reactivity of alternative halides, we additionally assessed bromide and chloride as leaving groups. Substituting bromobenzene (1a’) with 1a in the system resulted in a sluggish reaction (entry 18). Using chlorobenzene (1a”) as the reaction partner did not lead to any progress in the reaction (entry 19). Finally, decreasing the catalyst loading to 5 mol% produced comparable conversion with an 85% yield and 93% ee (entry 20).
|
|
Table 1 Reaction optimization.a |
{kind=link}
Having established the optimal conditions for the asymmetric reductive aryl addition, we proceeded to explore its applicability with various isatins, as depicted in Scheme 1. The reaction between iodobenzene (1b) and isatin 2a, lacking any substituents on its benzene core, yielded the product 2c in 66% yield and an impressive ee of 96%. The presence of methyl groups (3–4a) and isopropyl groups (5a) on the substituted isatins exhibited great compatibility with the reaction conditions. Similarly, the isatins 6–8a, endowed with OMe groups at various positions on the aryl rings, facilitated the formation of the desired products 6–8c with favorable yields and excellent enantioselectivities. Notably, isatins containing halogen substituents (F, Cl, and Br, 9–14a) demonstrated significant efficacy in the reductive 1,2-arylation, highlighting the potential value of this procedure alongside traditional cross-coupling reactions. Moreover, isatins carrying electron-withdrawing groups such as CF3 (15a), OCF3 (16–17a), and CO2Me (18a) displayed compatibility, affording the desired products 15–18c in moderate to good yields, accompanied by excellent enantioselectivities. It is worth noting the exemplary outcome achieved with the π-extended substrate of 1-methyl-1H-benzo[g]indole-2,3–dione (19a), which yielded the product 19c in a remarkable 93% yield and 94% ee. Furthermore, various N-substituted isatins, including trityl (20a), phenyl (21a), benzyl (22a), and p-methoxybenzyl (23a), were also well-tolerated within our system. Additionally, the compatibility of 5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline-1,2–dione (24a) with our reaction conditions was demonstrated, furnishing product 24c with outstanding enantiomeric excess. However, NH-free isatin 25a did not perform satisfactorily under the optimized reaction conditions.

|
Download:
|
| Scheme 1. Substrate scope of isatins. Reaction conditions: Co(dppf)Cl2 (10 mol%), (R,R)-L5 (10 mol%), 2–25a (0.10 mmol), 1b (0.14 mmol), B2nep2 (0.30 mmol), K3PO4 (0.30 mmol) in mesitylene (1 mL) at 85 ℃ for 24 h under N2 atmosphere. Isolated yields. ee values were determined by chiral HPLC. a Reacted for 48 h. | |
{kind=link}
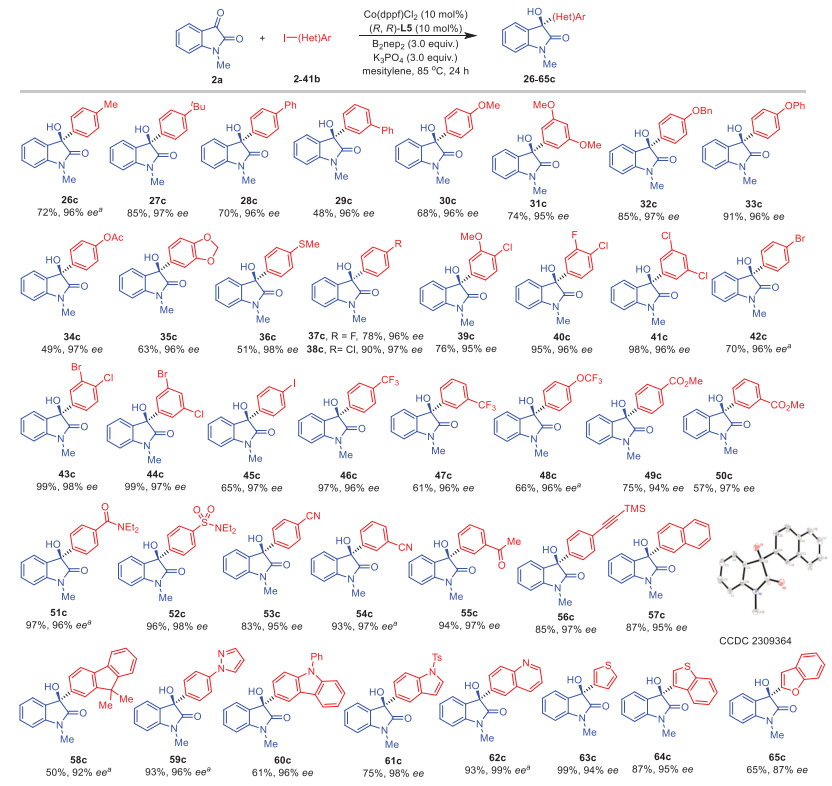
We conducted further investigation into the range of aryl and heteroaryl iodides (Scheme 2). Iodoarenes with diverse electron-neutral groups, such as methyl (2b), tert–butyl (3b), and phenyl (4–5b), yielded the corresponding products 26–29c with exceptional ee (>96%). Similarly, excellent enantioselectivities were observed for aryl iodides containing electron-donating groups, such as OMe (6–7b), OBn (8b), OPh (9b), OAc (10b), dioxole (11b), and SMe (12b), in the formation of products 30–36c. Notably, the presence of halogen groups including F, Cl, and Br (37–44c) had no impact on the reaction. Of particular interest is the usage of 1,4-diiodobenzene (21b), where one C-I bond could be preserved under the reaction conditions, resulting in the corresponding product 45c with a yield of 65% and an ee of 97%. Aryl iodides featuring electron-withdrawing substituents, such as trifluoromethyl (22–23b), trifluoromethoxy (24b), ester (25–26b), amide (27b), sulfonamide (28b), cyano (29–30b), and acetyl (31b), have generously bestowed upon us the resulting products 46–55c. These products have been graced with yields ranging from 57% to an impressive 97%, all the while maintaining ees exceeding the remarkable threshold of 95%. Moreover, the isatin derivative 32b with an ethynyl group could be effectively phenylated under these conditions. Using this strategy, polycyclic arenes such as 2-iodonaphthalene (33b) and 2-iodo-9,9-dimethyl-9H-fluorene (34b) could be employed with satisfactory enantioselectivities, and the absolute configuration of product 57c was confirmed via X-ray diffraction. Furthermore, the reaction displayed compatibility with a diverse array of heterocyclic patterns, encompassing pyrazole (35b), carbazole (36b), indole (37b), quinoline (38b), thiophene (39b), benzothiophene (40b), and benzofuran (41b), thereby yielding products 59–65c with varying yields ranging from 61% to 99% and impressive ee percentages ranging from 87% to 99%.

|
Download:
|
| Scheme 2. Substrate scope of aromatic iodides. Reaction conditions: Co(dppf)Cl2 (10 mol%), (R,R)-L5 (10 mol%), 2a (0.10 mmol), 2–41b (0.14 mmol), B2nep2 (0.30 mmol), K3PO4 (0.30 mmol) in mesitylene (1 mL) at 85 ℃ for 24 h under N2 atmosphere. Isolated yields; ee values were determined by chiral HPLC. aReacted for 48 h. | |
{kind=link}
In order to showcase the applicability of our approach, a sequence of trials was executed (Scheme 3). While NH-free isatin 25a was unsuccessful under the optimized reaction conditions, the removal of the trityl group in product 20c can be easily achieved under mild reaction conditions, resulting in the desired product 25c with excellent yield and enantioselectivity. Furthermore, we found that scaling up the reaction system did not have a significant impact on the product outcome. When the reaction of isatin 22a and PhI (1b) was carried out on a gram-scale, the product 22c was obtained with a yield of 76%, maintaining its efficiency. Additionally, the enantioselectivity of the reaction, with a 94% ee, was minimally affected as well. Furthermore, the chiral alcohol within the product can be further utilized in follow-up transformations including alkylation, acylation, and arylation, leading to the formation of products 66–68 with exceptional yields, while still maintaining excellent enantioselectivities.

|
Download:
|
| Scheme 3. Further investigations. Reaction conditions: (a) Et3SiH, TFA, DCM, 0 ℃ to r.t., 1 h; (b) MeI, NaH, DMF, 0 ℃ to r.t., 12 h; (c) Ac2O, DIPEA, 4-DMAP, DCM, r.t., 24 h; (d) PhB(OH)2, Cu(OAc)2, Et3N, 4-DMAP, 4 Å MS, DCM, under O2 atmosphere, r.t., 24 h. | |
{kind=link}
A sequence of mechanistic investigations was subsequently carried out in order to provide enlightenment into this reaction (Scheme 4). The arylboronate esters have the ability to easily engage in transmetallation with transition metals in carbonyl addition reactions [30,32-42]. To rule out the possibility of in situ formation of aryl boron species in the system, we initially examined the reactivity of Ar-Bnep (Scheme 4a). It was discovered that PhBnep (69) did not react with isatin 2a under the devised reaction conditions. Furthermore, iodoarene 70, bearing a Bnep motif, exhibited exceptional chemoselectivity as it exclusively yielded the desired product 71 in 63% yield with a remarkable ee (98%). This outcome dismisses the possiblity of in situ generation of the arylboron reagent for addition to the isatins. Subsequently, several intermolecular competition experiments were conducted to further scrutinize the reaction pathway (Scheme 4b). A competitive trial was conducted between isatins 6a and 17a, each featuring OMe and OCF3 groups respectively, yielding products 6c and 17c (17c/6c > 20/1). This finding establishes the intrinsic superiority of electron-deficient isatin in terms of reactivity, thereby providing evidence for a mechanism involving the nucleophilic addition pathway. In another competitive test, an equimolar mixture of aryl iodides 6b and 22b was used, resulting in a product ratio of 30c and 46c at 1/2.5. This indicates a higher reactivity of electron-withdrawing aryl iodides compared to the electron-rich ones. As depicted in Scheme 4c, the treatment of stoichiometric amounts of Co(dppf)Cl2 and ligand L5 led to the formation of uncoordinated dppf ligand and a new compound. The structure of this compound 72, further confirmed by X-ray diffraction, was determined to be a result of ligand exchange reactivity towards the background reaction, but demonstrate high activity after undergoing ligand exchange with L5. With a catalytic amount of compound 72 with B2nep2, product 1c was generated with a favorable outcome (96% yield and 97% ee). between Co(dppf)Cl2 and ligand L5. Additionally, when CoCl2 or Co(dppf)Cl2 was employed as the catalyst in the absence of L5, the formation of product 1c was not observed (Scheme 4d). These findings suggest that the cobalt sources examined do not display Therefore, this result implies that complex 72 functions as an intermediate in the reaction, while dppf does not actively participate in the catalytic cycle. Notably, replacing B2nep2 with B2pin2 in the reaction catalyzed by complex 72 resulted in a diminished outcome of product 1c (94% yield and 89% ee). This result, coupled with the findings from the reaction optimization experiments (Table 1, entries 9 and 10), highlights the bifunctional nature of boron species, which acts as not only a reducing agent, but also a determinant of enantioselectivity. Based on the complex 72, we have also obtained UV–visible absorption spectra of catalytic entities (Scheme 4e) [84-87]. The UV-visible spectra of compound 72 exhibited three conspicuous absorption peaks at 607, 659, and 712 nm. Upon reacting compound 72 with B2nep2, only a slight change in absorbance was observed, and subsequent addition of K3PO4 resulted in the vanishing of the characteristic peaks attributed to the Co(Ⅱ) complex. Simultaneously, a broad absorption band ranging from 367 nm to 525 nm appeared in the UV-visible spectra, signifying the formation of the Co(Ⅰ) complex [88-89]. Finally, a noticeable linear correlation was observed in the asymmetric reaction between isatin 1a and PhI (1b) in the presence of ligand L5 (Scheme 4f). This correlation implies that only one chiral ligand can coordinate with the cobalt center during the reaction [90-91].

|
Download:
|
| Scheme 4. Mechanistic experiments. | |
{kind=link}
Density functional theory (DFT) calculations were carried out to scrutinize the intricate mechanism of the reaction using compounds 1a and 2a as model substrates, as well as the determinants behind the observed high enantioselectivity and regioselectivity in the reaction (Fig. 2). Various Co-complexes with different spin states were preliminarily calculated, among which the spin states with a spin multiplicity of 3 for Co(Ⅰ) and Co(Ⅲ) and a spin multiplicity of 4 for Co(Ⅱ) are the lowest energy states (for details, see Supporting information). The initial step involves single-electron reduction of complex 72 by the boronate-base “ate” complex INT1A, generating a Co(Ⅰ) species INT2A with an endothermic energy of 6.4 kcal/mol. The giving intermediate INT2A undergoes σ-bond metathesis with the “ate” complex through transition state TS3A with an activation energy of 11.9 kcal/mol, resulting in the formation of active catalyst INT3A (Fig. 2a) [92,93]. Subsequently, the calculated energy profile for the catalytic cycle was depicted in Fig. 2b, where the Gibbs free energy of INT3A was set as the relative zero point. The oxidative addition of the C–I bond in substrate 2a to the low-valent cobalt center of INT3A, forming Co(Ⅲ) specie INT4A. This step is the rate-determining step of the catalytic cycle, with a surmountable energy barrier of 25.6 kcal/mol. The insertion of an isatin 1a can generate different Co(Ⅲ)-oxygen intermediates, and the regioselectivity is mainly determined by electronic factors. The natural population analysis (NPA) reveals that the β-carbonyl of 1a is more electron-rich compared to the α-carbonyl connected to the amino group, making it more favorable for the β-carbonyl to insert into the Co–C bond of INT4A. The energy barrier for carbonyl insertion leading to the formation of the α-selective product INT5B is 26.7 and 33.6 kcal/mol, respectively, which are higher than that for the formation of the β-selective product INT5A, suggesting that the formation of intermediate INT5A is kinetically favorable. The insertion of β-carbonyl can proceed through two different pathways, TS5A-R and TS5A-S, and the significant discrepancy in activation energy barriers between them is primarily attributed to the non-covalent interactions between the tBu group in chiral ligand, Bnep, and isatin. The independent gradient model based on Hirshfeld partition (IGMH) [94] shows that the favored transition state (TS5A-R) has stronger van der Waals interactions. In TS5A-R, the presence of three C–H…O hydrogen bonds (C–H1…O2 2.41 Å, C–H2…O1 2.36 Å, C–H3…O1 2.58 Å) facilitates the stabilization of TS5A-R and promotes the formation of the chiral intermediate INT5A-R. This calculational results align with the observed enantioselectivity, providing further theoretical evidence for the experimental findings. The preferred intermediate INT5A-R undergoes reduction elimination through the transition state TS6A-R with an activation energy of 1.5 kcal/mol, generating the R-configured product pro-R, which is then protonated to obtain the desired product (R)−3aa. Final reaction of the complex INT1A with intermediate INT6A regenerate the catalytic species INT3A through σ-bond metathesis with a calculated energy barrier of 4.7 kcal/mol, completing the catalytic cycle.

|
Download:
|
| Fig. 2. Proposed mechanism (DFT calculations were conducted at the M062X/def2TZVP/SMD(mesitylene)//B3LYP-D3BJ/def2SVP level of theory. | |
{kind=link}
In summary, we have successfully explored the utilization of cobalt catalysts to achieve enantioselective addition of aromatic halides to isatins using B2nep2 as the reducing agent. This strategy showcases remarkable compatibility with substrates containing sensitive functional groups, while obviating the need for organometallic reagents employed in prior methods. Considering the increasing significance of 3-aryl-3–hydroxy-2-oxindoles in the field of pharmacology, we envision that this technique will expedite the process of drug discovery and offer a valuable resource for the synthesis of these pharmacophores. Through a comprehensive integration of experimental and computational investigations, we have gained a profound comprehension of the reaction mechanism and the variables influencing the level of enantioselectivity.
Declaration of competing interestsThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementJieshuai Xiao: Validation, Methodology, Investigation. Yuan Zheng: Validation, Methodology, Investigation. Yue Zhao: Visualization. Zhuangzhi Shi: Writing – review & editing, Writing – original draft, Supervision. Minyan Wang: Writing – review & editing, Writing – original draft, Supervision.
AcknowledgmentsWe would like to thank the financial assistance provided by the National Key R&D Program of China (No. 2022YFA1503200), the National Natural Science Foundation of China (Nos. 22025104, 22171134, and 21972064), and the Fundamental Research Funds for the Central Universities (No. 020514380254) is greatly appreciated. We also extend our gratitude to the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University, for their generous support. Moreover, we would like to express our sincere appreciation to the High-Performance Computing Center of Nanjing University for performing the numerical calculations in this paper on its blade cluster system.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110243.
| [1] |
J. Bergman, J.O. Lindstrom, U. Tilstam, Tetrahedron 41 (1985) 2879-2881. DOI:10.1016/S0040-4020(01)96609-8 |
| [2] |
I. Chiyanzu, E. Hansell, J. Gut, et al., Bioorg. Med. Chem. Lett. 13 (2003) 3527-3530. DOI:10.1016/S0960-894X(03)00756-X |
| [3] |
C.V. Galliford, K.A. Scheidt, Angew. Chem. Int. Ed. 46 (2007) 8748-8758. DOI:10.1002/anie.200701342 |
| [4] |
B. Tan, N.R. Candeias, C.F. Barbas Ⅲ, Nat. Chem. 3 (2011) 473-477. DOI:10.1038/nchem.1039 |
| [5] |
L. Hong, R. Wang, Adv. Synth. Catal. 355 (2013) 1023-1052. DOI:10.1002/adsc.201200808 |
| [6] |
B.M. Trost, D.A. Bringley, T. Zhang, N. Cramer, J. Am. Chem. Soc. 135 (2013) 16720-16735. DOI:10.1021/ja409013m |
| [7] |
Y. Zhao, L. Liu, W. Sun, et al., J. Am. Chem. Soc. 135 (2013) 7223-7234. DOI:10.1021/ja3125417 |
| [8] |
B. Yu, D. Yu, H. Liu, Eur. J. Med. Chem. 97 (2015) 673-698. DOI:10.1016/j.ejmech.2014.06.056 |
| [9] |
B. Yu, Z. Yu, P.P. Qi, D.Q. Yu, H.M. Liu, Eur. J. Med. Chem. 95 (2015) 35-40. DOI:10.1016/j.ejmech.2015.03.020 |
| [10] |
G.S. Singh, Z.Y. Desta, Chem. Rev. 112 (2012) 6104-6155. DOI:10.1021/cr300135y |
| [11] |
R. Zeng, G. Dong, J. Am. Chem. Soc. 137 (2015) 1408-1411. DOI:10.1021/ja512306a |
| [12] |
X.C. Qiao, S.F. Zhu, Q.L. Zhou, Tetrahedron: Asymmetry 20 (2009) 1254-1261. DOI:10.1016/j.tetasy.2009.04.012 |
| [13] |
K. Zheng, C. Yin, X. Liu, et al., Angew. Chem. Int. Ed. 50 (2011) 2573-2577. DOI:10.1002/anie.201007145 |
| [14] |
S. Periyaraja, A.B. Mandal, P. Shanmugam, Org. Lett. 13 (2011) 4980-4983. DOI:10.1021/ol2022164 |
| [15] |
J. Guang, Q. Guo, J.C. Zhao, Org. Lett. 14 (2012) 3174-3177. DOI:10.1021/ol301270w |
| [16] |
G.W. Wang, A.X. Zhou, J.J. Wang, R.B. Hu, S.D. Yang, Org. Lett. 14 (2012) 2218-2221. DOI:10.1080/00103624.2012.701684 |
| [17] |
G.W. Wang, A.X. Zhou, J.J. Wang, R.B. Hu, S.D. Yang, Org. Lett. 15 (2013) 5270-5273. DOI:10.1021/ol402494e |
| [18] |
D. Cheng, Y. Ishihara, B. Tan, C.F. Barbas, ACS Catal. 4 (2014) 743-762. DOI:10.1021/cs401172r |
| [19] |
Y. Kuang, Y. Lu, Y. Tang, et al., Org. Lett. 16 (2014) 4244-4247. DOI:10.1021/ol501941n |
| [20] |
K. Pratap, A. Kumar, Org. Lett. 20 (2018) 7451-7454. DOI:10.1021/acs.orglett.8b03196 |
| [21] |
Y. Liu, Y.T. Xia, S.H. Cui, Y.G. Ji, L. Wu, Adv. Synth. Catal. 362 (2020) 2632-2636. DOI:10.1002/adsc.202000266 |
| [22] |
J. Khan, A. Tyagi, N. Yadav, R. Mahato, C.K. Hazra, J. Org. Chem. 86 (2021) 17833-17847. DOI:10.1021/acs.joc.1c02058 |
| [23] |
B. Yu, H. Xing, D.Q. Yu, H.M. Liu, Beilstein J. Org. Chem. 12 (2016) 1000-1039. DOI:10.3762/bjoc.12.98 |
| [24] |
S. Um, D.H. Bach, B. Shin, et al., Org. Lett. 18 (2016) 5792-5795. DOI:10.1021/acs.orglett.6b02555 |
| [25] |
T. Tokunaga, W.E. Hume, T. Umezome, et al., J. Med. Chem. 44 (2001) 4641-4649. DOI:10.1021/jm0103763 |
| [26] |
K.C. Nicolaou, M. Bella, D.Y.K. Chen, et al., Angew. Chem. Int. Ed. 41 (2002) 3495-3499. DOI:10.1002/1521-3773(20020916)41:18<3495::AID-ANIE3495>3.0.CO;2-7 |
| [27] |
A.W.G. Burgett, Q. Li, Q. Wei, P.G. Harran, Angew. Chem. Int. Ed. 42 (2003) 4961-4966. DOI:10.1002/anie.200352577 |
| [28] |
K.C. Nicolaou, M. Bella, D.Y.K. Chen, et al., J. Am. Chem. Soc. 126 (2004) 12888-12896. DOI:10.1021/ja040092i |
| [29] |
R.R. Knowles, J. Carpenter, S.B. Blakey, et al., Chem. Sci. 2 (2011) 308-311. DOI:10.1039/C0SC00577K |
| [30] |
R. Shintani, M. Inoue, T. Hayashi, Angew. Chem. Int. Ed. 45 (2006) 3353-3356. DOI:10.1002/anie.200600392 |
| [31] |
S. Lu, S.B. Poh, W.Y. Siau, Y. Zhao, Angew. Chem. Int. Ed. 52 (2013) 1731-1734. DOI:10.1002/anie.201209043 |
| [32] |
H. Lai, Z. Huang, Q. Wu, Y. Qin, J. Org. Chem. 74 (2009) 283-288. DOI:10.1021/jo802036m |
| [33] |
R. Shintani, K. Takatsu, T. Hayashi, Chem. Commun. 46 (2010) 6822-6824. DOI:10.1039/c0cc01635g |
| [34] |
J. Zhang, J. Chen, J. Ding, M. Liu, H. Wu, Tetrahedron 67 (2011) 9347-9351. DOI:10.1016/j.tet.2011.09.135 |
| [35] |
Z. Liu, P. Gu, M. Shi, P. McDowell, G. Li, Org. Lett. 13 (2011) 2314-2317. DOI:10.1021/ol200566s |
| [36] |
Y. Yamamoto, M. Yohda, T. Shirai, H. Ito, N. Miyaura, Chem. Asian J. 7 (2012) 2446-2449. DOI:10.1002/asia.201200481 |
| [37] |
J. Gui, G. Chen, P. Cao, J. Liao, Tetrahedron Asymmetry 23 (2012) 554-563. DOI:10.1016/j.tetasy.2012.04.013 |
| [38] |
Y. Zhuang, Y. He, Z. Zhou, et al., J. Org. Chem. 80 (2015) 6968-6975. DOI:10.1021/acs.joc.5b00595 |
| [39] |
J. Totobenazara, P. Bacalhau, A.A.S. Juan, et al., ChemistrySelect 1 (2016) 3580-3588. DOI:10.1002/slct.201600932 |
| [40] |
C.S. Marques, A.J. Burke, ChemCatChem 8 (2016) 3518-3526. DOI:10.1002/cctc.201600901 |
| [41] |
Y.Y. Zhang, H. Chen, X. Jiang, et al., Tetrahedron 74 (2018) 2245-2250. DOI:10.1016/j.tet.2018.03.039 |
| [42] |
W. Guo, S. Zhan, H. Yang, Z. Gu, Org. Lett. 25 (2023) 3281-3286. DOI:10.1021/acs.orglett.3c01028 |
| [43] |
D. Tomita, K. Yamatsugu, M. Kanai, M. Shibasaki, J. Am. Chem. Soc. 131 (2009) 6946-6948. DOI:10.1021/ja901995a |
| [44] |
X.A.F. Cook, A. de Gombert, J. McKnight, L.R.E. Pantaine, M.C. Willis, Angew. Chem. Int. Ed. 60 (2021) 11068-11091. DOI:10.1002/anie.202010631 |
| [45] |
Y. Okude, S. Hirano, T. Hiyama, H. Nozaki, J. Am. Chem. Soc. 99 (1977) 3179-3181. DOI:10.1021/ja00451a061 |
| [46] |
H. Jin, J. Uenishi, W.J. Christ, Y. Kishi, J. Am. Chem. Soc. 108 (1986) 5644-5646. DOI:10.1021/ja00278a057 |
| [47] |
D.A. Everson, D.J. Weix, J. Org. Chem. 79 (2014) 4793-4798. DOI:10.1021/jo500507s |
| [48] |
C.E.I. Knappke, S. Grupe, D. Gartner, et al., Chem. Eur. J. 20 (2014) 6828-6842. DOI:10.1002/chem.201402302 |
| [49] |
T. Moragas, A. Correa, R. Martin, Chem. Eur. J. 20 (2014) 8242-8258. DOI:10.1002/chem.201402509 |
| [50] |
D.J. Weix, Acc. Chem. Res. 48 (2015) 1767-1775. DOI:10.1021/acs.accounts.5b00057 |
| [51] |
J. Gu, X. Wang, W. Xue, H. Gong, Org. Chem. Front. 2 (2015) 1411-1421. DOI:10.1039/C5QO00224A |
| [52] |
K.E. Poremba, S.E. Dibrell, S.E. Reisman, ACS Catal. 10 (2020) 8237-8246. DOI:10.1021/acscatal.0c01842 |
| [53] |
W. Xue, X. Jia, X. Wang, et al., Chem. Soc. Rev. 50 (2021) 4162-4184. DOI:10.1039/d0cs01107j |
| [54] |
A. Duan, F. Xiao, Y. Lan, L. Niu, Chem. Soc. Rev. 51 (2022) 9986-10015. DOI:10.1039/d2cs00371f |
| [55] |
X. Pang, P.F. Su, X.Z. Shu, Acc. Chem. Res. 55 (2022) 2491-2509. DOI:10.1021/acs.accounts.2c00381 |
| [56] |
L. Xi, L. Du, Z. Shi, Chin. Chem. Lett. 33 (2022) 4287-4292. DOI:10.1016/j.cclet.2022.01.077 |
| [57] |
Y. Dai, F. Wang, S. Zhu, L. Chu, Chin. Chem. Lett. 33 (2022) 4074-4078. DOI:10.1016/j.cclet.2021.12.050 |
| [58] |
A. Nasim, G.T. Thomas, J.S. Ovens, et al., Org. Lett. 24 (2022) 7232-7236. DOI:10.1021/acs.orglett.2c03042 |
| [59] |
H. Chang, M. Jeganmohan, C. Cheng, Chem. Eur. J. 13 (2007) 4356-4363. DOI:10.1002/chem.200601880 |
| [60] |
H. Tao Jia, Q.S. Tian, J.N. Xiang, G.Z. Zhang, Chin. Chem. Lett. 28 (2017) 1182-1184. DOI:10.1016/j.cclet.2017.03.016 |
| [61] |
Z. Zhu, J. Xiao, M. Li, Z. Shi, Angew. Chem. Int. Ed. 61 (2022) e202201370. DOI:10.1002/anie.202201370 |
| [62] |
S. Zhang, S. Perveen, Y. Ouyang, et al., Angew. Chem. Int. Ed. 61 (2022) e202117843. DOI:10.1002/anie.202117843 |
| [63] |
X. Jiang, H. Jiang, Q. Yang, et al., J. Am. Chem. Soc. 144 (2022) 8347-8354. DOI:10.1021/jacs.2c02481 |
| [64] |
L. Zhang, M. Zhao, M. Pu, et al., J. Am. Chem. Soc. 144 (2022) 20249-20257. DOI:10.1021/jacs.2c05678 |
| [65] |
J. Xiao, M. Wang, X. Yin, et al., Angew. Chem. Int. Ed. 62 (2023) e202300743. DOI:10.1002/anie.202300743 |
| [66] |
H. Jiang, X.K. He, X. Jiang, et al., J. Am. Chem. Soc. 145 (2023) 6944-6952. DOI:10.1021/jacs.3c00462 |
| [67] |
L. Zhang, X. Wang, M. Pu, et al., J. Am. Chem. Soc. 145 (2023) 8498-8509. DOI:10.1021/jacs.3c00548 |
| [68] |
T. Liang, Y. Wu, J. Sun, et al., Chin. J. Chem. 41 (2023) 3253-3260. DOI:10.1002/cjoc.202300398 |
| [69] |
L.J. Li, Y. He, Y. Yang, et al., CCS Chem. 6 (2024) 537-584. DOI:10.31635/ccschem.023.202303412 |
| [70] |
C.H. Wei, S. Mannathan, C.H. Cheng, J. Am. Chem. Soc. 133 (2011) 6942-6944. DOI:10.1021/ja201827j |
| [71] |
T. Sawano, A. Ashouri, T. Nishimura, T. Hayashi, J. Am. Chem. Soc. 134 (2012) 18936-18939. DOI:10.1021/ja309756k |
| [72] |
K. Duvvuri, K.R. Dewese, M.M. Parsutkar, et al., J. Am. Chem. Soc. 141 (2019) 7365-7375. DOI:10.1021/jacs.8b13812 |
| [73] |
Y.L. Li, S.Q. Zhang, J. Chen, J.B. Xia, J. Am. Chem. Soc. 143 (2021) 7306-7313. DOI:10.1021/jacs.1c03527 |
| [74] |
K. Cui, Y.L. Li, G. Li, J.B. Xia, J. Am. Chem. Soc. 144 (2022) 23001-23009. DOI:10.1021/jacs.2c10021 |
| [75] |
P. Yang, Q. Wang, B.H. Cui, et al., J. Am. Chem. Soc. 144 (2022) 1087-1093. DOI:10.1021/jacs.1c11092 |
| [76] |
X. Jiang, W. Xiong, S. Deng, et al., Nat. Catal. 5 (2022) 788-797. DOI:10.1038/s41929-022-00831-1 |
| [77] |
A. Li, X. Song, Q. Ren, et al., Angew. Chem. Int. Ed. 62 (2023) e202301091. DOI:10.1002/anie.202301091 |
| [78] |
W. Xiong, X. Jiang, W.C. Wang, et al., J. Am. Chem. Soc. 145 (2023) 7983-7991. DOI:10.1021/jacs.2c13538 |
| [79] |
Z.L. Zhang, Z. Li, Y.T. Xu, et al., Angew. Chem. Int. Ed. 62 (2023) e202306381. DOI:10.1002/anie.202306381 |
| [80] |
W. Huang, J. Bai, Y. Guo, Chong Q, F. Meng, Angew. Chem. Int. Ed. 62 (2023) e202219257. DOI:10.1002/anie.202219257 |
| [81] |
Z. Liang, L. Wang, Y. Wang, et al., J. Am. Chem. Soc. 145 (2023) 3588-3598. DOI:10.1021/jacs.2c12475 |
| [82] |
H. Wang, X. Jie, Q. Chong, F. Meng, Nat. Commun. 15 (2024) 3427. DOI:10.1038/s41467-024-47719-1 |
| [83] |
L. Wang, Y. Tong, X. Lu, Y. Fu, Chin. Chem. Lett. 35 (2024) 109283. DOI:10.1016/j.cclet.2023.109283 |
| [84] |
M. Aresta, M. Rossi, A. Sacco, Inorg. Chim. Acta 3 (1969) 227-231. DOI:10.1016/S0020-1693(00)92484-8 |
| [85] |
K. Heinze, G. Huttner, L. Zsolnai, P. Schober, Inorg. Chem. 36 (1997) 5457-5469. DOI:10.1021/ic9705352 |
| [86] |
M. Gray, M.T. Hines, M.M. Parsutkar, et al., ACS Catal. 10 (2020) 4337-4348. DOI:10.1021/acscatal.9b05455 |
| [87] |
L. Wang, L. Wang, M. Li, Q. Chong, F. Meng, J. Am. Chem. Soc. 143 (2021) 12755-12765. DOI:10.1021/jacs.1c05690 |
| [88] |
M.R. Friedfeld, H. Zhong, R.T. Ruck, M. Shevlin, P.J. Chirik, Science 360 (2018) 888-893. DOI:10.1126/science.aar6117 |
| [89] |
H. Zhong, M.R. Friedfeld, P.J. Chirik, Angew. Chem. Int. Ed. 58 (2019) 9194-9198. DOI:10.1002/anie.201903766 |
| [90] |
D. Guillaneux, S. Zhao, O. Samuel, D. Rainford, H.B. Kagan, J. Am. Chem. Soc. 116 (1994) 9430-9439. DOI:10.1021/ja00100a004 |
| [91] |
C. Girard, H.B. Kagan, Angew. Chem. Int. Ed. 37 (1998) 2922-2959. DOI:10.1002/(SICI)1521-3773(19981116)37:21<2922::AID-ANIE2922>3.0.CO;2-1 |
| [92] |
X. Lu, Y. Wang, B. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 12632-12637. DOI:10.1021/jacs.7b06469 |
| [93] |
J. Sheng, H.Q. Ni, S.X. Ni, et al., Angew. Chem. Int. Ed. 60 (2021) 15020-15027. DOI:10.1002/anie.202102481 |
| [94] |
T. Lu, Q. Chen, J. Comput. Chem. 43 (2022) 539-555. DOI:10.1002/jcc.26812 |