 2025, Vol. 36
2025, Vol. 36 


The incorporation of fluorine atoms into organic molecules can profoundly alter their chemical and pharmacokinetic attributes, making it a subject of increasing interest among both academic and industrial organic chemists [1-8]. Specifically, the catalytic formation of C(sp3)-F bonds, especially with precise stereoselective control, has garnered significant attention. Over the past several decades, many elegant methodologies have emerged to access these coveted stereocenters [9-15]. Nonetheless, many of these approaches are limited to creating sp3-F stereocenters adjacent to functional groups like carbonyls and benzyls. The catalytic assembly of α-fluoro stereogenic carbon centers remote to a functional group, utilizing readily available starting materials, remains an elusive yet imperative objective in synthetic organic chemistry [16-18].
Hydrofluorination of alkenes, one of the most abundant feedstocks in chemical synthesis, offers an exceptionally straightforward approach for expeditiously forging aliphatic fluorides, including those distal to a functional group [19]. Significant achievements have been made in this area using electrophilic or nucleophilic fluorinating agents. However, catalytic enantioselective hydrofluorination of alkenes to access α-fluoro-substituted carbon stereogenic centers remains underexploited [20-25]. Two elegant asymmetric protocols (Scheme 1a) were independently disclosed by the groups of MacMillan [20] and Huang [22]. In these reactions, enals can undergo hydrofluorination with high levels of enantioselectivity by using chiral organocatalysts. Nonetheless, these methods restrict the use of α,β-unsaturated aldehydes as alkene partners. In this regard, transition metal-catalyzed hydrofluorination with metal hydrides [26-28] presents a promising avenue to broaden the scope of accessible alkenes. For instance, the Gouverneur group developed the first example of hydrofluorination of stryenes utilizing silanes and electrophilic fluorination reagents, yielding regioselective benzylic fluorides [21]. This chemistry involves the migratory insertion of a palladium hydride into styrene to form an alkyl-metal species, which then reacts with an electrophilic fluorinating agent via oxidative addition/reductive elimination. The stereoselectivity of newly formed C-F bonds can be controlled using chiral ligands, as demonstrated by Gouverneur, and further developed by Liao and Lei (Scheme 1b) [21,23]. Recently, an alternative pathway involving a metal-hydride hydrogen atom transfer (MHAT) process [27,29-31] has emerged as a promising strategy for facilitating hydrofluorination of alkenes. In this process, hydrogen atom transfer from a metal hydride to an alkene generates an alkyl radical, which subsequently abstracts a fluorine atom from an electrophilic fluorinating agent or metal-fluoro species, leading to the formation of alkyl fluorides, as pioneered by Boger and coworkers using iron hydrides (Scheme 1b) [32-36]. While the MHAT strategy generally shows good compatibility of functional groups, enhancing its synthetic utility, the establishment of an enantioselective variant, particularly for unactivated internal alkenes, remains a longstanding challenge. Recently, the groups of Chu and Yu developed the first radical transfer strategy for the asymmetric hydrogenation of alkenyl fluorides utilizing nickel hydride hydrogen atom transfer [37]. To our knowledge, no examples of MHAT-enabled asymmetric hydrofluorination of alkenes have yet been disclosed.

|
Download:
|
| Scheme 1. Asymmetric hydrofluorination of internal alkenes via radical mechanism. | |
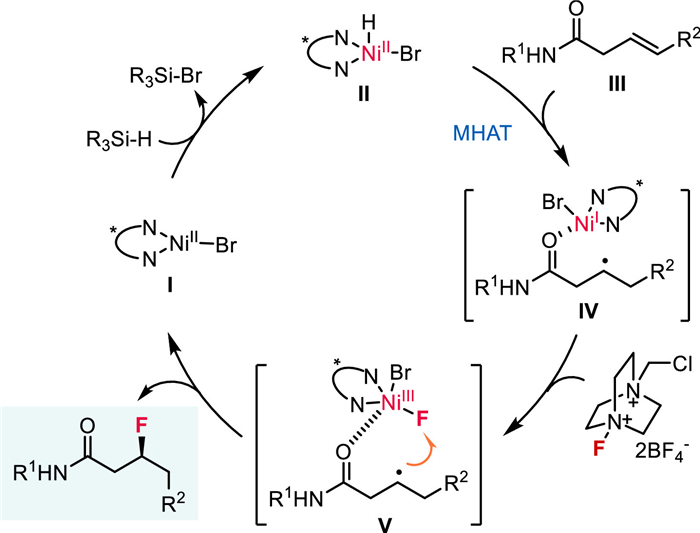
In connection with our interest in nickel-catalyzed hydro- and di-functionalization reactions of alkenes and alkynes [37-45], we questioned whether a combination of NiH–HAT [34,37,46] and fluorine atom transfer would lead to an asymmetric hydrofluorination of alkenes (Scheme 1c). The proposed mechanism is depicted in Scheme 2. The reaction begins with the reaction of ligated Ni(Ⅱ)Br Ⅰ with silane to form Ni(Ⅱ)-H species Ⅱ. With the weak coordination of amide group, Ni(Ⅱ)-H Ⅱ undergoes regioselective HAT with alkene to generate a solvent-caged radical pair Ⅳ. Then, the reaction of Ⅳ with Selectfluor produces a Ni(Ⅲ)-fluoro species Ⅴ, followed by an outer-sphere fluorine atom transfer to the alkyl radical, delivering the desired hydrofluorination product and Ni(Ⅱ)Br to close the cycle. Specifically, we envisaged using a well-designed chiral nickel complex effectively controlling the stereoselectivity of either the NiH–HAT or the radical fluorine transfer process. Drawing from our recent investigations into asymmetric hydrogenation of alkenes [37], we were optimistic about the feasibility of NiH–HAT with stereoselective control. In terms of the fluorine atom transfer, transition metal-catalyzed C(sp3)-F bond formation reactions, entailing the transfer of fluorine atoms from metal-fluoro species to alkyl radicals, have been extensively explored by Groves [47,48], Li [49-51], and others [52-57]. More recently, an asymmetric radical fluorine transfer catalyzed by iron enzymes has been disclosed [57]. Critically, the carbon-based radical generated via MHAT is commonly perceived as a solvent-caged radical pair [31], thereby conferring advantages to the stereoselective control of fluorine atom transfer from Ni-fluoro to alkyl radical, owing to spatial proximity [57]. To address the regioselectivity, another inherent challenge associated with catalytic hydrofunctionalization of unactivated internal alkenes [58-62], we anticipated that the use of weakly coordinating functional groups such as amides, as previously reported by our group [38,40] and others [63-65], would facilitate the control of regioselectivity via chelation. Herein, we report the development of a nickel-catalyzed highly regio- and enantioselective hydrofluorination of internal alkenes by merging NiH–HAT and fluorine transfer processes (Scheme 1c). These reactions demonstrate a broad substrate scope with excellent regio- and enantioselectivity, delivering optically pure β-F amides under mild conditions.

|
Download:
|
| Scheme 2. Proposed mechanism. | |
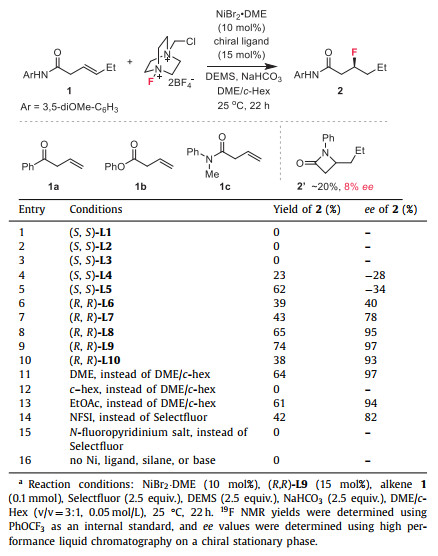
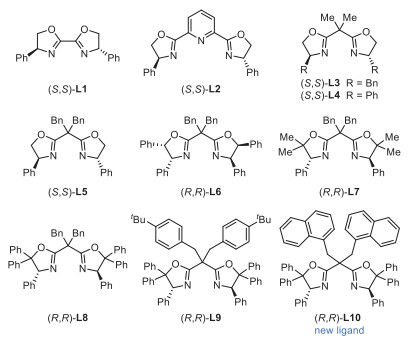
Initial investigations on a template internal alkene 1 revealed that the reactions of 1 with Selectfluor, diethoxymethylsilane (DEMS), and NaHCO3 in the presence of catalytic NiBr2·DME with chiral bi-oxazoline (BiOx) L1, pyridine-2,6-bis(oxazolines) (Pybox) L2, or Bn-substituted bisoxazoline (Box) L3 failed to afford any desired products (Fig. 1, Table 1, entries 1–3). Switching to the Ph-Box L4 gave a 23% yield of the desired β-F amide product 2 with exclusive regioselectivity and a promising enantiomeric excess (entry 4). Encouraged by these results, we further evaluated a series of chiral Ph-Box ligands (L5-L10). We found that an increase in the steric hindrance at the gem‑disubstituted linker or the C5 substituents of Box ligands can improve the efficiency of this hydrofluorination reaction (entries 5–10). Remarkably, the C5-diphenyl substituted ligands (R,R)-L8 and (R,R)-L9 with bulky linkers significantly improved the yields and ee, and (R,R)-L9 proved to be optimal, affording product 2 in 74% yield and 97% ee (entries 8 and 9). Further increasing the steric hindrance of the linker by installing two naphthalene methyl groups gave a new ligand (R,R)-L10, nevertheless resulting in a dramatic decrease in yields (entry 10). As anticipated, the choice of solvent proved to be pivotal. Reactions conducted in DME or ethyl acetate yielded moderate yields, whereas no product was observed in cyclohexane (entries 11–13). The use of N-fluorobenzenesulfonimide (NFSI) led to significant decreases in yields and ee; nevertheless, no product was detected with N-fluoropyridinium salt as a fluorinating agent (entries 14 and 15). While almost concurrently, Hong reported the nickel-catalyzed asymmetric hydrofluorination of unactivated alkenes utilizing (S,S)-L3 and a N-fluoropyridinium salt via an ionic pathway. However, attempts to react alkenes with (S,S)-L3/Selectfluor or (R,R)-L9/N-fluoropyridinium salt were unsuccessful (entries 3 and 15), indicating that different mechanisms are influenced by the backbone of chiral ligands and F+ agents [66]. Control experiments revealed that nickel catalyst, chiral ligand, silane, and base are all necessary for this transformation (entry 16). We identified the β-CONH group is crucial for efficiency and regio-/stereo-selectivity, as no products were observed in cases involving alkenyl ketone (1a), alkenyl ester (1b), or tertiary amide (1c). Additionally, alongside product 2, a small amount of side product lactam 2′ was detected in most cases.

|
Download:
|
| Fig. 1. Structures of chiral ligands tried in this study. | |
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
{kind=link}
{kind=link}
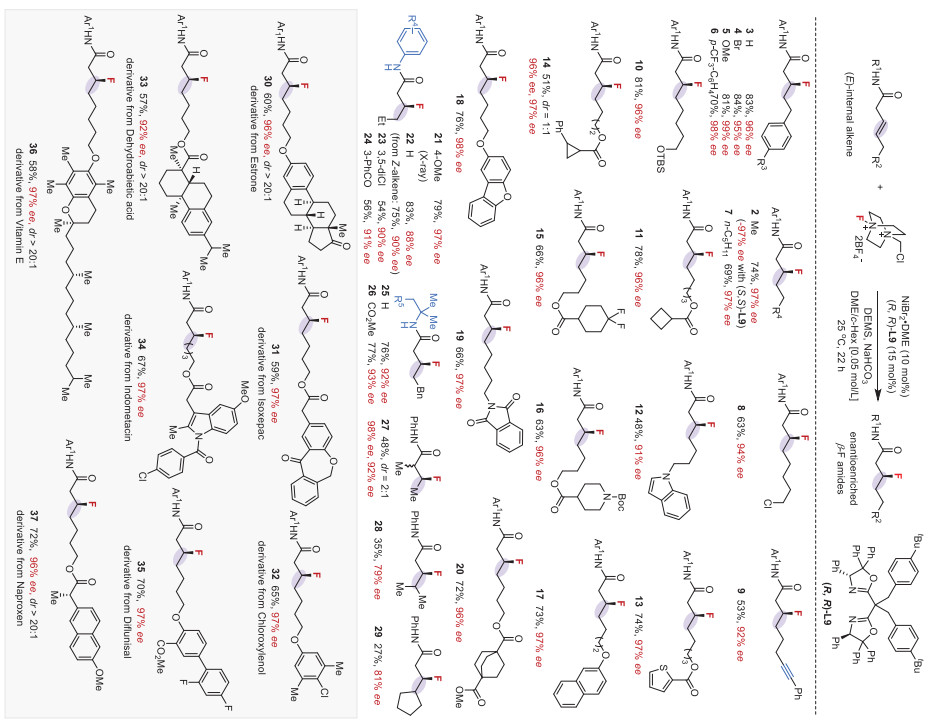
We then explored the internal alkenes under optimal conditions (Scheme 3). β,γ−Alkenyl amides tethered with benzyls, alkyls, internal alkynes, ethers, esters, heterocycles, and imides exhibited good reactivity, yielding the corresponding β-fluoro amide products with exceptional enantioselectivities (3–29, up to 99% ee). The mild conditions are compatible with many important functional groups, including fluoride-sensitive and acid-sensitive groups such as silyl ether and Boc. The electronic properties of the N-aromatic ring of alkenyl amides showed some impact on the efficiency and enantioselectivity, and substrates with electron-poor aryl-protected amides proceeded with slightly decreased yields and ee (23–24). N-Alkyl substituted alkenyl amides were suitable coupling partners, affording the corresponding chiral aliphatic fluorides with good yields and high ee (25–26). The reaction of α-methyl substituted alkenyl amide proceeded with good enantioselectivity, albeit with a significantly decreased yield (27, 2:1 dr, 98% ee, 92% ee). Furthermore, the reactions of γ,γ-disubstituted alkenyl amides formed β-fluoro amides with excellent regioselectivity, albeit with decreased ee and low yields (28–29). The absolute configuration of the newly formed sp3-F chiral center was established by X-ray crystallographic analysis (21).

|
Download:
|
| Scheme 3. Asymmetric hydrofluorination of internal alkenes. Reaction conditions: alkene (0.1 mmol), NiBr2·DME (10 mol%), (R,R)-L9 (15 mol%), Selectfluor (2.5 equiv.), DEMS (2.5 equiv.), NaHCO3 (2.5 equiv.), DME/c-Hex (v/v = 3:1, 0.05 mol/L), 25 ℃, 22 h. Isolated yields; ee values were determined using high performance liquid chromatography on a chiral stationary phase. (E)-Alkenes were used if otherwise denoted. | |
{kind=link}
To showcase the versatility and applicability of this methodology, we carried several complex internal alkenes derived from biologically active agents for late-stage asymmetric hydrofluorination. As shown in Scheme 3, internal alkenes tethered with drugs and agrochemicals, including estrone, isoxepac, chloroxylenol, dehydroabietic acid, indomethacin, diflunisal, vitamin E, and naproxen, all proceeded smoothly, delivering the enantioenriched alkyl fluorides with moderate yields and excellent ee (30–37, 92%−97% ee).
Next, we sought to perform mechanistic studies to probe potential pathways (Scheme 4). First, the use of pre-prepared (R,R)-L8-NiBr2 complex [67] Ni-I in place of NiBr2·dme/(R,R)-L8 delivered product 2 in comparable yield and ee (Scheme 4a). A non-linear relationship between the enantiopurities of product 2 and ligand L9 was observed, further supporting that a mono-nickel complex with a single chiral ligand is involved in this transformation (Scheme 4b). A key step for the proposed mechanism involves MHAT to alkene forming a carbon radical/nickel(Ⅱ) cage pair. To probe the potential involvement of radical intermediacy, we subjected radical inhibitor TEMPO (1.0 equiv.) to the reaction system, which completely shut down the hydrofluorination reaction (Scheme 4c). Furthermore, the reaction of diene 1e under the optimal condition gave a 39% yield of cycloisomerization products 38 (a mixture of regioisomers and diastereoisomers), suggesting the involvement of alkyl radicals (Scheme 4d) [68]; no fluorination product was observed, probably due to the sluggish fluorine atom transfer to alkyl radicals without the directing effect of amides. On the other hand, the reaction of cyclic alkene 1f gave a 32% yield of product 39 in a mixture of diastereoisomers (dr = 2:1) (Scheme 4e); a similar result was obtained in the case of product 27 (dr = 2:1) shown in Scheme 3. The low dr ratios are in sharp contrast to the previous report by Martin and coworkers [69], wherein excellent diastereoselectivity was observed for the β-C-C bond formation via an amide-coordinated nickelacyle species, indicating that an outer-sphere ligand transfer pathway is likely involved for the C-F bond-forming in our system. Next, the reaction of alkene 1d and Ph2SiD2 showed a kinetic isotope effect (KIE) of 1.25 on the initial rates of the reaction, consistent with our previous NiH–HAT work (Scheme 4f) [37]. To further identify the stereo-determining process, the reactions of internal alkenes (E)- and (Z)−1d with Ph2SiD2 were conducted; product 22-d was obtained with low diastereomeric ratios (1.4:1 and 3.2:1, respectively), suggesting that the C(sp3)-F bond formation, presumably via fluorine atom transfer from nickel-fluoro species, could be the stereo-determining step (Scheme 4g). Ongoing studies aim to provide further support for the proposed Ni-HAT/fluorine atom transfer process.

|
Download:
|
| Scheme 4. Mechanistic studies. | |
{kind=link}
In conclusion, we have developed an efficient method for the asymmetric hydrofluorination of internal alkenes with silane and Selectfluor via a NiH–HAT/fluorine atom transfer process. A wide array of chiral β-fluoro amides can be obtained from abundant internal alkenes with very high levels of regioselectivity and enantioselectivity at room temperature. This methodology demonstrates a broad substrate generality and tolerates a wide range of functional groups, including electron-rich heterocycles, silyl ethers, and Boc. The transformation described herein serves as a valuable addition to the repertoire of methods for the enantioselective synthesis of β-fluoro substituted stereogenic carbon centers at remote positions and, more importantly, stimulates further exploration in the area of asymmetric fluorine atom transfer reactions.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementFan Chen: Methodology, Investigation, Formal analysis, Data curation. Xiaoyu Zhao: Investigation. Weihang Miao: Investigation, Data curation. Yingying Li: Investigation. Ye Yuan: Investigation. Lingling Chu: Writing – review & editing, Visualization, Validation, Supervision, Resources, Project administration, Funding acquisition, Formal analysis, Data curation, Conceptualization.
AcknowledgmentsThis research was made possible as a result of a generous grant from the Fundamental Research Funds for the Central Universities (Nos. 2232024Y-01, 2232024A-03) and the National Science Fund for Excellent Young Scholars (No. 22122101). We thank the staffs from the College of Materials Science and Engineering at Donghua University for X-ray data collection using Brucker D8 Venture.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110239.
| [1] |
K. Müller, C. Faeh, F. Diederich, Science 317 (2007) 1881-1886. DOI:10.1126/science.1131943 |
| [2] |
D. O’Hagan, Chem. Soc. Rev. 37 (2008) 308-319. DOI:10.1039/B711844A |
| [3] |
S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320-330. DOI:10.1039/B610213C |
| [4] |
M. Inoue, Y. Sumii, N. Shibata, ACS Omega 5 (2020) 10633-10640. DOI:10.1021/acsomega.0c00830 |
| [5] |
H. Mei, A.M. Remete, Y. Zou, et al., Chin. Chem. Lett. 31 (2020) 2401-2413. DOI:10.1016/j.cclet.2020.03.050 |
| [6] |
Q. Wang, H. Song, Q. Wang, Chin. Chem. Lett. 33 (2022) 626-642. DOI:10.1016/j.cclet.2021.07.064 |
| [7] |
J. He, Z. Li, G. Dhawan, et al., Chin. Chem. Lett. 34 (2023) 107578. DOI:10.1016/j.cclet.2022.06.001 |
| [8] |
Q. Wang, Y. Bian, G. Dhawan, et al., Chin. Chem. Lett. 35 (2024) 109780. DOI:10.1016/j.cclet.2024.109780 |
| [9] |
V.A. Brunet, D. O’Hagan, Angew. Chem. Int. Ed. 47 (2008) 1179-1182. DOI:10.1002/anie.200704700 |
| [10] |
J.R. Wolstenhulme, V. Gouverneur, Acc. Chem. Res. 47 (2014) 3560-3570. DOI:10.1021/ar500282z |
| [11] |
X. Yang, T. Wu, R.J. Phipps, F.D. Toste, Chem. Rev. 115 (2015) 826-870. DOI:10.1021/cr500277b |
| [12] |
P.A. Champagne, J. Desroches, J.D. Hamel, M. Vandamme, J.F. Paquin, Chem. Rev. 115 (2015) 9073-9174. DOI:10.1021/cr500706a |
| [13] |
Y. Zhu, J. Han, J. Wang, et al., Chem. Rev. 118 (2018) 3887-3964. DOI:10.1021/acs.chemrev.7b00778 |
| [14] |
I.N.M. Leibler, S.S. Gandhi, M.A. Tekle Smith, A.G. Doyle, J. Am. Chem. Soc. 145 (2023) 9928-9950. DOI:10.1021/jacs.3c01824 |
| [15] |
C. Chen, L. Fu, P. Chen, G. Liu, Chin. J. Chem. 35 (2017) 1781-1788. DOI:10.1002/cjoc.201700489 |
| [16] |
V. Rauniyar, A.D. Lackner, G.L. Hamilton, F.D. Toste, Science 334 (2011) 1681-1684. DOI:10.1126/science.1213918 |
| [17] |
N.A. Cochrane, H. Nguyen, M.R. Gagne, J. Am. Chem. Soc. 135 (2013) 628-631. DOI:10.1021/ja3116795 |
| [18] |
C. Sandford, R. Rasappan, V.K. Aggarwal, J. Am. Chem. Soc. 137 (2015) 10100-10103. DOI:10.1021/jacs.5b05848 |
| [19] |
X. Bertrand, L. Chabaud, J.F. Paquin, Chem. Asian J. 16 (2021) 563-574. DOI:10.1002/asia.202001403 |
| [20] |
Y. Huang, A.M. Walji, C.H. Larsen, D.W.C. MacMillan, J. Am. Chem. Soc. 127 (2005) 15051-15053. DOI:10.1021/ja055545d |
| [21] |
E. Emer, L. Pfeifer, J.M. Brown, V. Gouverneur, Angew. Chem. Int. Ed. 53 (2014) 4181-4185. DOI:10.1002/anie.201310056 |
| [22] |
L. Wang, X. Jiang, J. Chen, Y. Huang, Angew. Chem. Int. Ed. 58 (2019) 7410-7414. DOI:10.1002/anie.201902989 |
| [23] |
X. Yin, B. Chen, F. Qiu, et al., ACS Catal. 10 (2020) 1954-1960. DOI:10.1021/acscatal.9b05264 |
| [24] |
H. Egami, Y. Hamashima, Chem. Rec. 23 (2023) e202200285. DOI:10.1002/tcr.202200285 |
| [25] |
E.A. McKnight, D. Cadwallader, C.M. Le, Eur. J. Org. Chem. 27 (2023) e202300017. |
| [26] |
A.J. Jordan, G. Lalic, J.P. Sadighi, Chem. Rev. 116 (2016) 8318-8372. DOI:10.1021/acs.chemrev.6b00366 |
| [27] |
S.W.M. Crossley, C. Obradors, R.M. Martinez, R.A. Shenvi, Chem. Rev. 116 (2016) 8912-9000. DOI:10.1021/acs.chemrev.6b00334 |
| [28] |
N.A. Eberhardt, H. Guan, Chem. Rev. 116 (2016) 8373-8426. DOI:10.1021/acs.chemrev.6b00259 |
| [29] |
R.W. Hoffmann, Chem. Soc. Rev. 45 (2016) 577-583. DOI:10.1039/C5CS00423C |
| [30] |
S.A. Green, S.W.M. Crossley, J.L.M. Matos, et al., Acc. Chem. Res. 51 (2018) 2628-2640. DOI:10.1021/acs.accounts.8b00337 |
| [31] |
S.L. Shevick, C.V. Wilson, S. Kotesova, et al., Chem. Sci. 11 (2020) 12401-12422. DOI:10.1039/d0sc04112b |
| [32] |
T.J. Barker, D.L. Boger, J. Am. Chem. Soc. 134 (2012) 13588-13591. DOI:10.1021/ja3063716 |
| [33] |
H. Shigehisa, E. Nishi, M. Fujisawa, K. Hiroya, Org. Lett. 15 (2013) 5158-5161. DOI:10.1021/ol402696h |
| [34] |
P. Song, S. Zhu, ACS Catal. 10 (2020) 13165-13170. DOI:10.1021/acscatal.0c03884 |
| [35] |
Y. Xie, P.W. Sun, Y. Li, et al., Angew. Chem. Int. Ed. 58 (2019) 7097-7101. DOI:10.1002/anie.201902607 |
| [36] |
J. Liu, J. Rong, D.P. Wood, et al., J. Am. Chem. Soc. 146 (2024) 4380-4392. DOI:10.1021/jacs.3c10989 |
| [37] |
F. Chen, Q. Zhang, Y. Li, Z.X. Yu, L. Chu, J. Am. Chem. Soc. 146 (2024) 11418-11431. |
| [38] |
H.Y. Tu, F. Wang, L. Huo, et al., J. Am. Chem. Soc. 142 (2020) 9604-9611. DOI:10.1021/jacs.0c03708 |
| [39] |
S. Zhu, X. Zhao, H. Li, L. Chu, Chem. Soc. Rev. 50 (2021) 10836-10856. DOI:10.1039/d1cs00399b |
| [40] |
F. Wang, S. Pan, S. Zhu, L. Chu, ACS Catal. 12 (2022) 9779-9789. DOI:10.1021/acscatal.2c02163 |
| [41] |
Y. Dai, F. Wang, S. Zhu, L. Chu, Chin. Chem. Lett. 33 (2022) 4074-4078. DOI:10.1016/j.cclet.2021.12.050 |
| [42] |
L. Huo, X. Li, Y. Zhao, L. Li, L. Chu, J. Am. Chem. Soc. 145 (2023) 9876-9885. DOI:10.1021/jacs.3c02748 |
| [43] |
X. Li, M. Yuan, F. Chen, et al., Chem 9 (2023) 154-169. DOI:10.1016/j.chempr.2022.09.020 |
| [44] |
S. Pan, F. Chen, Y. Zhang, L. Shao, L. Chu, Angew. Chem. Int. Ed. 62 (2023) e202305426. DOI:10.1002/anie.202305426 |
| [45] |
Y. Fan, Z. Huang, Y. Lu, S. Zhu, L. Chu, Angew. Chem. Int. Ed. 63 (2024) e202315974. DOI:10.1002/anie.202315974 |
| [46] |
C. Yao, S. Wang, J. Norton, M. Hammond, J. Am. Chem. Soc. 142 (2020) 4793-4799. DOI:10.1021/jacs.9b13757 |
| [47] |
W. Liu, X. Huang, M.J. Cheng, et al., Science 337 (2012) 1322-1325. DOI:10.1126/science.1222327 |
| [48] |
W. Liu, J.T. Groves, Angew. Chem. Int. Ed. 52 (2013) 6024-6027. DOI:10.1002/anie.201301097 |
| [49] |
F. Yin, Z. Wang, Z. Li, C. Li, J. Am. Chem. Soc. 134 (2012) 10401-10404. DOI:10.1021/ja3048255 |
| [50] |
Z. Liu, H. Chen, Y. Lv, et al., J. Am. Chem. Soc. 140 (2018) 6169-6175. DOI:10.1021/jacs.8b03077 |
| [51] |
L. Zhu, C. Li, Transition metal-mediated radical fluorination for preparing alkyl fluorides, in: J. Hu, T. Umemoto (Eds.), Fluorination, Springer Singapore, Singapore, 2018, pp. 1–17.
|
| [52] |
H. Zhao, X. Fan, J. Yu, C. Zhu, J. Am. Chem. Soc. 137 (2015) 3490-3493. DOI:10.1021/jacs.5b00939 |
| [53] |
J.K. Bower, A.D. Cypcar, B. Henriquez, S.C.E. Stieber, S. Zhang, J. Am. Chem. Soc. 142 (2020) 8514-8521. DOI:10.1021/jacs.0c02583 |
| [54] |
H. Hintz, J. Bower, J. Tang, et al., Chem. Catal. 3 (2023) 100491. DOI:10.1016/j.checat.2022.100491 |
| [55] |
C. Panda, O. Anny-Nzekwue, L.M. Doyle, R. Gericke, A.R. McDonald, JACS Au 3 (2023) 919-928. DOI:10.1021/jacsau.3c00021 |
| [56] |
H. Zhang, X. Sun, C. Ma, et al., ACS Catal. 14 (2024) 3115-3127. DOI:10.1021/acscatal.3c05154 |
| [57] |
Q. Zhao, Z. Chen, J. Soler, et al., Nat. Synth. 3 (2024) 958-966. DOI:10.1038/s44160-024-00507-7 |
| [58] |
R.I. McDonald, G. Liu, S.S. Stahl, Chem. Rev. 111 (2011) 2981-3019. DOI:10.1021/cr100371y |
| [59] |
H. Wen, G. Liu, Z. Huang, Coord. Chem. Rev. 386 (2019) 138-153. DOI:10.1016/j.ccr.2019.01.024 |
| [60] |
J. Guo, Z. Cheng, J. Chen, X. Chen, Z. Lu, Acc. Chem. Res. 54 (2021) 2701-2716. DOI:10.1021/acs.accounts.1c00212 |
| [61] |
W. Zhao, H.X. Lu, W.W. Zhang, B.J. Li, Acc. Chem. Res. 56 (2023) 308-321. DOI:10.1021/acs.accounts.2c00713 |
| [62] |
Y. Li, X. Lu, Y. Fu, CCS Chem. 6 (2024) 1130-1156. DOI:10.31635/ccschem.024.202303678 |
| [63] |
J. Derosa, R. Kleinmans, V.T. Tran, et al., J. Am. Chem. Soc. 140 (2018) 17878-17883. DOI:10.1021/jacs.8b11942 |
| [64] |
W. Zhao, K.Z. Chen, A.Z. Li, B.J. Li, J. Am. Chem. Soc. 144 (2022) 13071-13078. DOI:10.1021/jacs.2c05993 |
| [65] |
H.J. Kang, C. Lee, S. Hong, Angew. Chem. Int. Ed. 62 (2023) e202305042. DOI:10.1002/anie.202305042 |
| [66] |
C. Lee, M. Kim, S. Han, D. Kim, S. Hong, J. Am. Chem. Soc. 146 (2024) 9375-9384. DOI:10.1021/jacs.4c01548 |
| [67] |
Z. Wang, H. Yin, G.C. Fu, Nature 563 (2018) 379-383. DOI:10.1038/s41586-018-0669-y |
| [68] |
S.W.M. Crossley, F. Barabé, R.A. Shenvi, J. Am. Chem. Soc. 136 (2014) 16788-16791. DOI:10.1021/ja5105602 |
| [69] |
J. Rodrigalvarez, H. Wang, R. Martin, J. Am. Chem. Soc. 145 (2023) 3869-3874. DOI:10.1021/jacs.2c12915 |