 2025, Vol. 36
2025, Vol. 36 


b University of Chinese Academy of Sciences, Beijing 100049, China;
c Institute of Molecular Science, Shanxi University, Taiyuan 030006, China;
d College of Smart Energy, Shanghai Jiao Tong University, Shanghai 200240, China
Carbon dioxide (CO2) is an attractive C1 source for the construction of fine chemicals and valuable drug products. The catalytic C–C coupling with CO2 has currently received considerable attention from the scientific community [1-4]. However, the activation and utilization of CO2 are still problematic due to its inherent thermodynamic stability and/or kinetic inertness. Many transition metals based catalytic systems have been developed for selective CO2 transformation [5-12].
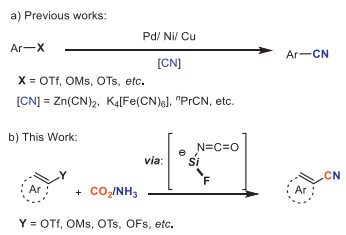
On the other hand, nitriles are key synthons used in the synthesis of aldehydes, ketones, carboxylic acids, alcohols, amides, amines, and heterocycles [13-15]. In the past decades, preparation of cyano-containing compounds using transition metal catalysts developed rapidly. And the direct employment of metal/metalloid-bound cyanides or the in situ generation of cyanide ion, such as KCN [16-18], Zn(CN)2 [19-21], TMSCN [22,23], acetone cyanohydrin [24,25], is often involved. Higher activity was demonstrated by these cyanide reagents. Nonetheless, catalyst deactivation was usually observed because of the fast liberation of cyanide ligands, causing a limited substrate scope [26-28].
In this regard, the general cyanation of aryl triflates is rarely investigated [29] while phenol analogues exist extensively in bulk chemicals, natural products and pharmaceuticals. In 2010, Kwong et al. [30] reported palladium-catalyzed cyanation of aryl mesylates. Here, only very limited examples were suitable for utilizing K4[Fe(CN)6] due to the heterogeneous nature of the reaction mixture. To address these concerns, Morandi [31] employed butyronitrile as a cyanide (CN) source to achieve nickel-catalyzed cyanation of aryl triflates. And different non-metallic cyano-group sources including acetonitrile, 2-methyl-2-phenylmalononitrile (MPMN) and morpholinoacetonitrile [32-35] were then utilized in transition metal catalyzed cyanation reaction of phenol analogues (Scheme 1a).

|
Download:
|
| Scheme 1. Catalytic cyanations of C(sp2)–O bond with different cyano sources. | |
Meanwhile, peculiar fluoride effect has already been proved critical on the carboxylation with CO2 [36,37], seeming mainly to stabilize intermediates. Based on the above reports and the continuation of our interest on the catalytic cyanation using CO2 [38,39], we report here the cyanation of C–O bonds with CO2 and NH3 starting from the investigation of fluoride effect (Scheme 1b).
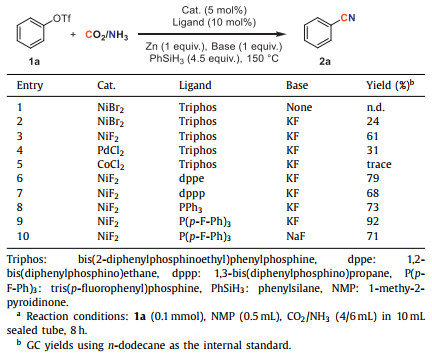
Phenyl trifluoromethanesulfonate 1a was chosen as the model substrate to investigate catalytic cyanation with CO2 and NH3 (Table 1). Firstly, KF was demonstrated exceptional efficacy in this transformation since the target product 2a was not detected without F source. And 24% yield of 2a could be obtained when KF was added (entry 2). Various metal catalysts were then examined (entries 2–5). NiF2 was the optimal choice, likely due to favorable C–O bond insertion facilitated by the small anion radius of fluorine. Surprisingly, evaluation of ligands indicated that simple PPh3 provided an almost comparable yield of 73% compared to bidentate or multidentate ligands (entries 6–8). It was recognized that electron-deficient phosphine ligands exhibited somewhat distinctive effects on the C–C coupling reaction [40-44]. And a similar phenomenon was noted in our system. Tris(p-fluorophenyl)phosphine (P(p-F-Ph)3) was found to be superior to other ligands, yielding 2a with 92% yield (entry 9). And a modest yield was obtained when NaF was used instead of KF (entry 10).
|
|
Table 1 Optimization of reaction conditions. |
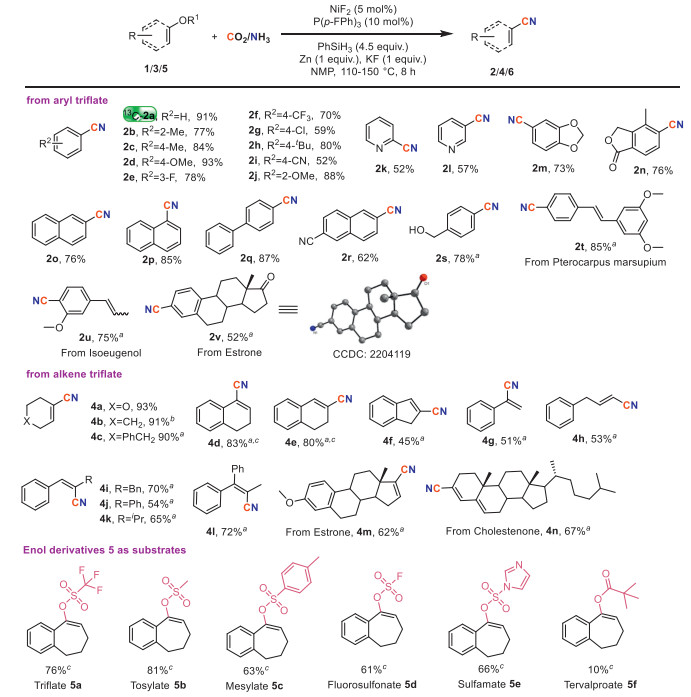
Based on the encouraging results mentioned above, the versatility and applicability of this system were further investigated under the optimized conditions, as depicted in Scheme 2. An array of substrates bearing electron-donating groups (alkyl, OMe, naphthyl) as well as electron-withdrawing groups (F, Cl, CF3, CN) underwent the cyanation reaction smoothly. 13C-labeled nitrile 13C-2a was conveniently obtained with 13CO2 (91% yield). And triflates incorporating a series of different heterocyclic patterns, including benzodioxole, phthalide, and pyridine were tested and the products 2k-2n were isolated with high yields. In addition, the cyanation of polycyclic compounds 1o-1p took place as expected, affording the corresponding arylnitriles with satisfactory yields (76%–85%). It is noteworthy that alcohol group was also tolerated under the system, providing the desired nitrile 2s in 78% yields. We then proceeded to assess the applicability in modifying bioactive intermediates. Specifically, cyanation of pterocarpus marsupium derivative 1t provided (E)-4-(3,5-dimethoxystyryl)benzonitrile 2t in excellent yield (85%). Isoeugenol derivative was successfully cyanated to produce 2u in good yield (75%). And the functionalized nitrile 2v could be readily prepared from estrone derivative, with 52% isolated yield attained.

|
Download:
|
| Scheme 2. Reaction conditions: substrate (0.1 mmol), CO2/NH3 (4/6 mL) in 10 mL sealed tube, NMP (0.5 mL) at 150 ℃ for 8 h. Isolated yield. a dppe instead of P(p-F-Ph)3. b NMR yield with 1,1,2,2-tetrachloroethane as internal standard. c 110 ℃. | |
Vinyl triflates were found compatible, allowing access to important synthon α,β-unsaturated nitriles. A series of substrates exhibiting different electronic properties reacted delicately to furnish desired products 4a-4n in moderate to excellent yields (45%–93%). For example, oxyheterocyclohexene triflate 3a was converted to 4a in 93% yield. Cyanated cyclohexene ring products 4b and 4c were both obtained with high yields. Cycloalkenes triflates 3d-3f resulted in similarly acceptable yields (45%–83%). Cyanation of external olefin 3g gave medium yield (51%). When linear vinyl triflate 3h was used as the substrate, cyanated product 4h was formed as the major product. It was found that sterically hindered substituent on α-position of the C–O bond gave rise to relatively low yields 4i-4k. And the reaction also occurred smoothly when trisubstituted alkene substrate was employed, providing cyanation product 4l in decent yield (72%). Moreover, derivatives 3m-3n from steroid hormones estrone and cholestenone were successfully cyanated, resulting in corresponding nitriles 4m and 4n with yields of 62% and 67%, respectively. These results not only highlighted the preferable performance of the catalytic system in accomplishing cyanation but also demonstrated the generality of this vinyl triflates cyanation reaction. Furthermore, the cyanation of other enol derivatives 5a-5f with different o-protecting groups were investigated under the optimized conditions. It was found that alcohols substituted with OTf (5a), OMs (5b), OTs (5c) as well as OFs (5d) underwent the cyanation reaction smoothly. In addition, vinyl imidazole sulfonate 5e was a suitable coupling partner. And, vinyl trimethylacetate 5f was also be compatible with this catalytic system, albeit with a low yield of 10%.
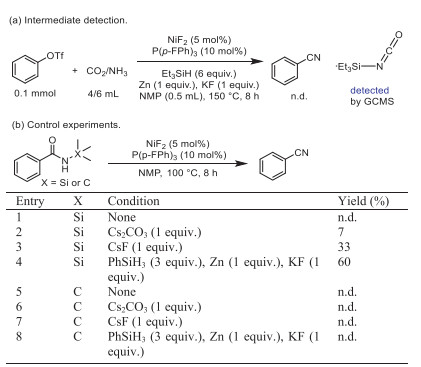
Control experiments were devised and conducted to gain insight into this transformation. In the presence of Et3SiH, no nitrile product 2a was observed while triethylsilyl isocyanate could be detected by GCMS (Scheme 3a and Fig. S3 in Supporting information). So, the formation of active silyl isocyanates were considered as the possible intermediate in this reaction [45]. In addition, the use of amidate metal as the reaction intermediate has been extensively investigated, particularly in the synthesis and conversion of amides [46-51]. In our prior research, we observed that amides could be efficiently converted to nitriles using HBpin [52]. Consequently, we hypothesized that the coupling and deoxygenation processes involving isocyanate could potentially be achieved through synergistic silane/nickel catalysis mediated by amidate metal intermediates. N-Silyl benzamides were prepared as substrates to test the feasibility of this proposal (Scheme 3b). No cyanated product was detected with nickel and ligand only (Scheme 3b, entry 1). And benzonitrile was formed in modest yield in the presence of cesium salt (entries 2 and 3). These results indicated that the alkali metal promoted the deoxygenation of amide and F− served as the accelerator via Si-F interaction. Surprisingly, the yield of benzonitrile can reach 60% under similar standard conditions (entry 4). In contrast, no benzonitrile was formed under the same conditions when Si in N-silyl benzamides was replaced by C (Scheme 3b, entries 5–8). These results imply that the cyanation through an in situ formation and transformation of silyl amide moiety is feasible.

|
Download:
|
| Scheme 3. Mechanistic probe for the reaction. (a) Intermediate detection. (b) Control experiments. Reaction scale (0.1 mmol), NMP (0.5 mL). | |
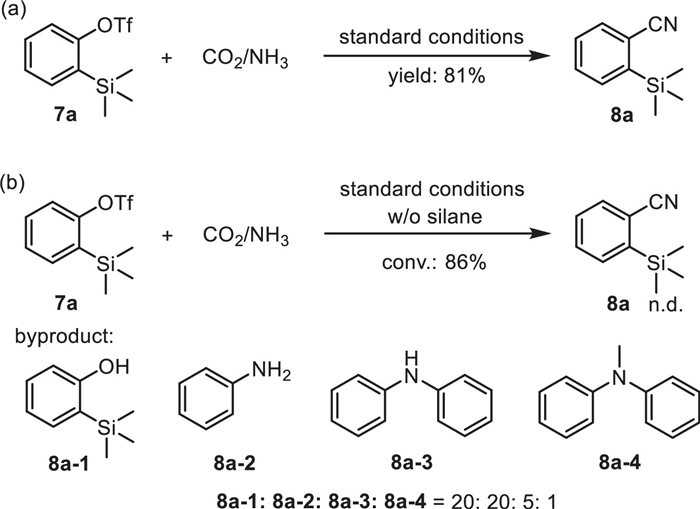
Given the importance of Si-containing nitriles, we then turned attention to exploring cyanation of the substrates with silyl group on the basis of our discovery. o-Silylaryl triflates, recognized as Kobayashi’s precursor, have been extensively employed in aryne chemistry due to their ready availability and facile generation of arynes through fluoride-induced 1,2-elimination [53,54]. Therefore, we propose precise control of chemoselectivity through the fluorine-silicon chelation with o-silylaryl triflate. To our delight, nitrile product 8a was formed in 81% yield under standard conditions (Scheme 4a). Then, cyanation of 7a without silane was conducted. No desired product was detected despite the high conversion while arylamines were the main byproducts, which indicated the formation of arynes from 7a due to the activation of fluoride. And hydrosilanes might play an important role in this system, as they could not only act as reduction reagents but also serve as accelerating agents by promoting the cleavage of C–OTf bond basing on the high fluorophilicity [55-59]. Thus, aryl triflates with silyl substituent were also tolerant.

|
Download:
|
| Scheme 4. Cyanidation of o-silylaryl triflate 7a. | |
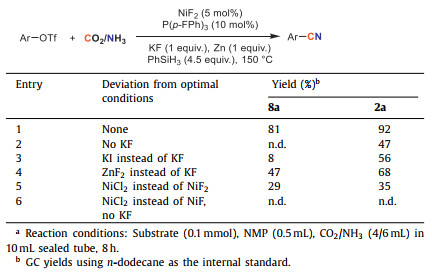
To demonstrate the key role of Si–F interaction, a series of control experiments were conducted (Table 2). When 7a used as the substrate, desired product 8a was not detected under otherwise identical conditions but without KF (entry 2). Only 8% yield was obtained with KI instead of KF while ZnF2 delivering 8a in 47% yield (entries 3 and 4). And NiCl2 instead of NiF2 as the catalyst yields the cyanation product in obviously reduced yield. No product was detected without F source (entry 6). These results revealed that fluoride plays a crucial role in the reaction pathway. Similar results were observed when 1a was used as the substrate. Standard conditions resulted in a satisfactory reaction with 92% yield. Visibly lower yield of 2a was detected without KF. And ZnF2 furnished 2a in an acceptable yield (entry 4, 68%) while NiCl2 was less reactive (entry 5, 35%). No target product was detected without F source as well (entry 6). These results implied that Si–F interaction also has a promoting effect on the cyanation for substrates.
|
|
Table 2 Control experiments for F-Si interaction. |
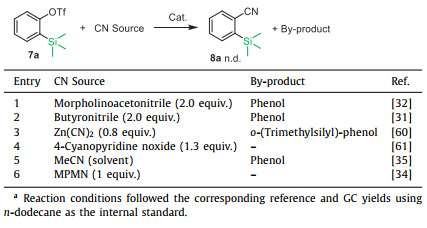
A series of comparative experiments were then conducted using other established Ni-catalyzed cyanation systems utilizing various cyano sources (Table 3). The optimized reaction conditions reported in the literature were directly employed without alteration. None of these previously known catalytic systems yielded the desired cyanation product, underscoring the distinct advantages of our approach. Partial breakage of the C–Si bond in the substrate occurred due to hydrodesilylation during the reaction process. Additionally, phenol was occasionally detected due to the hydrolysis of triflates.
|
|
Table 3 Results by using the conditions of the reported systems.a |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
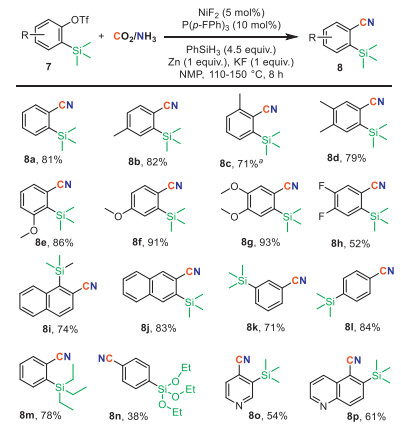
We further explored the scope of this cyanation reaction with a variety of silylaryl triflate (Scheme 5). Gratifyingly, a variety of aryne precursors bearing diverse substituents were well tolerated, yielding the corresponding products 8a-8p with moderate to good yields. Functional groups such as ethers 7e-7g, fluorides 7h, and naphthalene 7i-7j were compatible. Substrates bearing TMS groups at the meta- or para-position of the C–O bond 7k-7l were also amenable to this transformation, affording the respective nitriles 8k and 8l in good yields. Aryl triflates with triethylsilyl 7m and triethylsiloxy groups 7n resulted in moderate yields, with benzonitrile detected as the main by-product derived from hydrodesilylation. Heterocycle-containing substrates such as pyridine 7o and quinolone 7p also exhibited favorable reactivity, providing heteroaryl nitriles in comparable yields.

|
Download:
|
| Scheme 5. Reaction conditions: substrate (0.1 mmol), CO2/NH3 (4/6 mL) in 10 mL sealed tube, NMP (0.5 mL) at 150 ℃ for 8 h. Isolated yield. a dppe instead of P(p-F-Ph)3. | |
{kind=link}
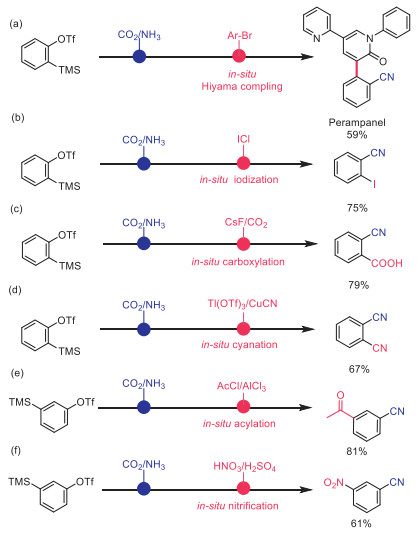
Since organosilicon compounds have been found to be versatile motif in organic synthesis and materials science, this catalytic system provides a good opportunity for fortifying the cyano-containing products. Perampanel (brand name: Fycompa), which is the first noncompetitive AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid) receptor antagonist authorized by FDA [62-64], could be conveniently obtained via Pd-catalyzed Hiyama reaction [65-67] with a synthetically useful yield (Scheme 6a). Then sequential cyanation and iodination was realized by adding iodine monochloride [68,69], affording 75% yield of 2-cyanophenyliodobenzene (Scheme 6b). The optimized conditions of carboxylation reported in the literature were applied without modification [70-72]. And, satisfactory selectivity for benzoic acid was achieved (Scheme 6c). In addition, phthalonitrile could also be obtained with modest yield (67%) by using the reported method (Scheme 6d) [73-75]. Furthermore, well known acylation or nitration methods could be employed directly, and the corresponding products were attained with acceptable yields (81% and 61% respectively).

|
Download:
|
| Scheme 6. Synthetic utility. Conditions: (a) CuF2 (10 mol%), Pd2(dba)3 (1.0 mol%), tris(2,4,6-trimethoxyphenyl)phosphine (4.0 mol%), CsF (1.2 equiv.) DMI, 120 ℃, 17 h. (b) ICl (2.2 equiv.), CHCl3, reflux, 6 h. (c) CsF (1 equiv.), CO2 (1 atm.), DMF, 60 ℃, 1 h. (d) Tl(OTf)3 (1.1 equiv.), CuCN (4 equiv.), MeCN, reflux, 17 h. (e) AcCl (2 equiv.), AlCl3 (2 equiv.), CS2, room temperature, 7 h. (f) HNO3, H2SO4, 0 ℃ to room temperature. | |
{kind=link}
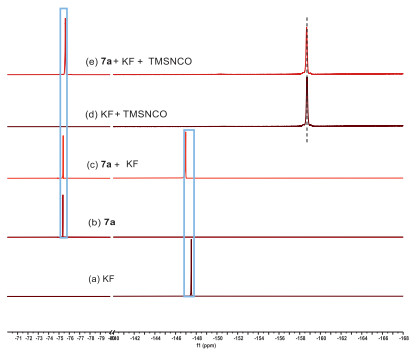
To elucidate the Si-F interaction modes in this transformation, a series of 19F NMR experiments were conducted (Scheme 7). All chemical shifts were recorded at 298 K in methanol unless otherwise specified. Notably, the chemical shift value of KF shifted significantly upfield upon addition of TMS-NCO (trimethylsilylisocyanate) (Schemes 7a and d). This outcome indicates the presence of a bonding-like effect between the fluorine (in KF) and silicon (in TMS-NCO), which was further supported by the 29Si NMR spectra of TMS-NCO (Figs. S7a and d in Supporting information, downfield). HRMS of the mixture of TMS-NCO and KF was carried out at room temperature, and a peak at m/z = 134.0434 was observed and assigned to their adduct. A slight downfield peak of KF was observed (Schemes 7a–c) when exposed to substrate 7a, attributed to a static inductive effect. Furthermore, when 7a, KF, and TMS-NCO were combined, both KF and 7a exhibited upfield shifts in chemical shift (Scheme 7e), accompanied by a notable downfield shift in the 29Si NMR spectrum of TMS-NCO (Fig. S7e in Supporting information). These shifts in chemical shift values suggest the emergence of a bonding-like effect between the fluorine (in KF) and silicon (in TMS-NCO) through transient pentacoordinated silicon anion species, while the interaction between fluorine (in KF) and silicon (in 7a) is likely governed by a spatial electrostatic effect.

|
Download:
|
| Scheme 7. 19F NMR experiments. | |
{kind=link}
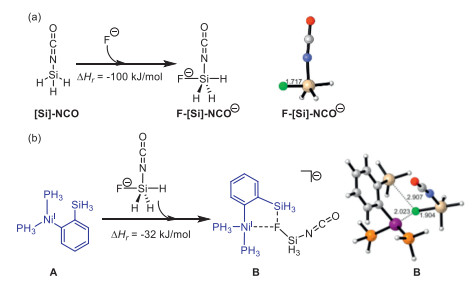
Besides, DFT calculations were carried out to understand the important promotion effect of F-Si interaction in this work (Scheme 8). As a simplification, we used PH3 and SiH3 to model the P(p-F-Ph)3 ligand and SiMe3 group, respectively. The optimized 3D structure was displayed by CYLview program. Scheme 8a shows that the reaction between F− and silyl isocyanate, forming a pentacoordinate Si adduct, has a reaction enthalpy of −100 kJ/mol, indicating that F− effectively stabilizes the highly reactive silyl isocyanate species. Furthermore, the interaction of pentacoordinate anionic silyl isocyanate F-[Si]-NCO− with Ni complex of A to form complex B has a reaction enthalpy of −32 kJ/mol, highlighting the strong electrostatic interactions between F− and Ni as well as the SiH3 of A with the F–Ni and F–Si distance of 2.023 and 2.907 Å, respectively (Scheme 8b).

|
Download:
|
| Scheme 8. (a) The reaction between F− and silyl isocyanate with formation of F-[Si]-NCO. (b) The interaction of complex A and F-[Si]-NCO−. | |
{kind=link}
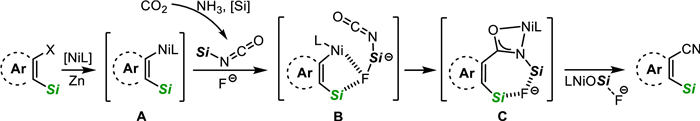
Based on both experimental results and literature reports [76], the reaction pathway for o-silylaryl triflate is proposed, as illustrated in Scheme 9. Initially, nucleophilic Ni(Ⅰ) species A is formed in the presence of zinc powder and silane (refer to Supporting information). Simultaneously, the fluoro-silicon adduct is produced through the capture of in-situ generated silyl isocyanate from CO2 and NH3 by fluoride, owing to its robust affinity. This adduct is subsequently attracted by the intrinsic silyl group in species A, leading to the formation of five membered ring species B. Then, the nickel-carbon insertion process, involving an intramolecular-like nucleophilic attack, occurs readily. Consequently, amidate nickel species C is formed, accompanied by changes in the bonding strength between F and Si. Further transformation leads to the desired product through a conceivable silyl N-to-O migration, releasing the fluoride-attached nickel silanoxy.

|
Download:
|
| Scheme 9. Proposed reaction pathway for o-silylaryl triflate. | |
{kind=link}
In summary, we have developed an innovative approach for the nickel-catalyzed reductive cyanation of aryl pseudohalides, utilizing CO2 and NH3 as the carbon and nitrogen sources, respectively. This system exhibits a wide substrate scope with excellent tolerance to various functional groups. We successfully implemented selective relay bifunctionalization of silyl-containing compounds, including aryne precursors. The F-Si interactions might play dual roles: 1) formation of the penta-coordinated fluoro-silicon adduct through the capture of in-situ generated silyl isocyanate by fluoride; 2) for substrates with vicinal silyl group, further making coupling step kinetically favorable due to the inherent affinity between F and Si. Through a carefully crafted Si–F–Si interaction model, we achieved excellent chemo-selectivity by modulating the strength of the F-Si bond. Furthermore, the practical utility of this catalytic system was demonstrated through the modification of biologically active compounds.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementYang Li: Writing – review & editing, Writing – original draft, Visualization, Methodology, Investigation. Yanan Dong: Writing – original draft, Funding acquisition, Validation, Supervision, Investigation, Conceptualization. Zhihong Wei: Writing – original draft, Supervision, Software. Changzeng Yan: Supervision, Resources, Project administration. Zhen Li: Data curation, Writing – review & editing. Lin He: Resources, Project administration, Funding acquisition. Yuehui Li: Writing – original draft, Supervision, Resources, Project administration, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (Nos. 22072167, 22202218) and the Jiangsu Natural Science Funds for Young Scholar (No. BK20211093) is greatly appreciated.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110206.
| [1] |
T. Sakakura, J.C. Choi, H. Yasuda, Chem. Rev. 107 (2007) 2365-2387. DOI:10.1021/cr068357u |
| [2] |
C.D.N. Gomes, et al., Angew. Chem. Int. Ed. 51 (2012) 187-190. DOI:10.1002/anie.201105516 |
| [3] |
J. Artz, et al., Chem. Rev. 118 (2018) 434-504. DOI:10.1021/acs.chemrev.7b00435 |
| [4] |
M. Aresta, A. Dibenedetto, Dalton Trans. 28 (2007) 2975-2992. DOI:10.1039/b700658f |
| [5] |
G. Bertuzzi, A. Cerveri, L. Lombardi, et al., Chin. J. Chem. 39 (2021) 3116-3126. DOI:10.1002/cjoc.202100450 |
| [6] |
J.H. Ye, T. Ju, H. Huang, et al., Acc. Chem. Res. 54 (2021) 2518-2531. DOI:10.1021/acs.accounts.1c00135 |
| [7] |
L. Wang, C. Qi, W. Xiong, et al., Chin. J. Catal. 43 (2022) 1598-1617. DOI:10.1016/S1872-2067(21)64029-9 |
| [8] |
H. Luo, J. Ren, Y. Sun, et al., Chin. Chem. Lett. 34 (2023) 107782. DOI:10.1016/j.cclet.2022.107782 |
| [9] |
Y.Y. Gui, S.S. Yan, W. Wang, et al., Sci. Bull. 68 (2023) 3124-3128. DOI:10.1016/j.scib.2023.11.018 |
| [10] |
C.K. Ran, H.Z. Xiao, L.L. Liao, et al., Natl. Sci. Open 2 (2023) 20220024. DOI:10.1360/nso/20220024 |
| [11] |
A. Tortajada, F. Juliá-Hernández, M. Börjesson, et al., Angew. Chem. Int. Ed. 57 (2018) 15948-15982. DOI:10.1002/anie.201803186 |
| [12] |
Q. Liu, L. Wu, R. Jackstell, M. Beller, Nat. Commun. 6 (2015) 5933. DOI:10.1038/ncomms6933 |
| [13] |
Z. Rappoport, Chemistry of The Cyano Group. London: John Wiley & Sons, 1970.
|
| [14] |
A.J. Fatiadi, S. Patai, Z. Rappoport, Preparation and Synthetic Applications of Cyano Compounds. New York: Wiley-VCH, 1983.
|
| [15] |
A. Kleemann, J. Engel, B. Kutscher, et al., Pharmaceutical Substance: Synthesis, Patents and Applications of The Most Relevant APIs. fifth ed.. Stuttgart: Thieme, 2008.
|
| [16] |
K. Takagi, T. Okamoto, S. Yasumasa, et al., Bull. Chem. Soc. Jpn. 48 (1975) 3298-3301. DOI:10.1246/bcsj.48.3298 |
| [17] |
M.R.I. Chambers, D.A. Widdowson, J. Chem. Soc., Perkin Transac. 1 7 (1989) 1365-1366. |
| [18] |
K. Takagi, Y. Sakakibara, Chem. Lett. 18 (1989) 1957-1958. DOI:10.1246/cl.1989.1957 |
| [19] |
D.M. Tschaen, R. Desmond, A.O. King, et al., Synth. Commun. 24 (1994) 887-890. DOI:10.1080/00397919408011310 |
| [20] |
F.G. Buono, R. Chidambaram, R.H. Mueller, R.E. Waltermire, Org. Lett. 10 (2008) 5325-5328. DOI:10.1021/ol8016935 |
| [21] |
P. Ryberg, Org. Process Res. Dev. 12 (2008) 540-543. DOI:10.1021/op800020r |
| [22] |
H. Nakamura, H. Shibata, Y. Yamamoto, Tetrahedron Lett. 41 (2000) 2911-2914. DOI:10.1016/S0040-4039(00)00284-7 |
| [23] |
M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg, M. Beller, J. Organomet. Chem. 684 (2003) 50-55. DOI:10.1016/S0022-328X(03)00503-5 |
| [24] |
M. Sundermeier, A. Zapf, M. Beller, Angew. Chem. Int. Ed. 42 (2003) 1661-1664. DOI:10.1002/anie.200250778 |
| [25] |
F. Burg, J. Egger, J. Deutsch, N. Guimond, Org. Process Res. Dev. 20 (2016) 1540-1545. DOI:10.1021/acs.oprd.6b00229 |
| [26] |
P. Anbarasan, T. Schareina, M. Beller, Chem. Soc. Rev. 40 (2011) 5049-5067. DOI:10.1039/c1cs15004a |
| [27] |
P.J. Amal Joseph, S. Priyadarshini, Org. Process Res. Dev. 21 (2017) 1889-1924. DOI:10.1021/acs.oprd.7b00285 |
| [28] |
G. Yan, Y. Zhang, J. Wang, Adv. Synth. Catal. 359 (2017) 4068-4105. DOI:10.1002/adsc.201700875 |
| [29] |
B.M. Rosen, K.W. Quasdorf, D.A. Wilson, et al., Chem. Rev. 111 (2011) 1346-1416. DOI:10.1021/cr100259t |
| [30] |
P.Y. Yeung, C.M. So, C.P. Lau, F.Y. Kwong, Angew. Chem. Int. Ed. 49 (2010) 8918-8922. DOI:10.1002/anie.201005121 |
| [31] |
P. Yu, B. Morandi, Angew. Chem. Int. Ed. 56 (2017) 15693-15697. DOI:10.1002/anie.201707517 |
| [32] |
R. Takise, K. Itami, J. Yamaguchi, Org. Lett. 18 (2016) 4428-4431. DOI:10.1021/acs.orglett.6b02265 |
| [33] |
A. Chatupheeraphat, H.H. Liao, S.C. Lee, M. Rueping, Org. Lett. 19 (2017) 4255-4258. DOI:10.1021/acs.orglett.7b01905 |
| [34] |
L.R. Mills, J.M. Graham, P. Patel, S.A.L. Rousseaux, J. Am. Chem. Soc. 141 (2019) 19257-19262. DOI:10.1021/jacs.9b11208 |
| [35] |
Y. Ueda, N. Tsujimoto, T. Yurino, et al., Chem. Sci. 10 (2019) 994-999. DOI:10.1039/c8sc04437f |
| [36] |
T.V.Q. Nguyen, W.J. Yoo, S. Kobayashi, Asian J. Org. Chem. 7 (2018) 116-118. DOI:10.1002/ajoc.201700519 |
| [37] |
S. Ichii, G. Hamasaka, Y. Uozumi, Chem. Asian J. 14 (2019) 3850-3854. DOI:10.1002/asia.201901155 |
| [38] |
H. Wang, Y. Dong, C. Zheng, et al., Chem 4 (2018) 2883-2893. DOI:10.1016/j.chempr.2018.09.009 |
| [39] |
Y. Dong, P. Yang, S. Zhao, Y. Li, Nat. Commun. 11 (2020) 4096. DOI:10.1038/s41467-020-17939-2 |
| [40] |
W.R. Moser, C.J. Papile, D.A. Brannon, et al., J. Mol. Catal. 41 (1987) 271-292. DOI:10.1016/0304-5102(87)80106-2 |
| [41] |
C.P. Casey, E.L. Pausen, E.W. Beuttenmueller, et al., J. Am. Chem. Soc. 119 (1997) 11817-11825. DOI:10.1021/ja9719440 |
| [42] |
A.M. Banet Osuna, W. Chen, E.G. Hope, et al., J. Chem. Soc., Dalton Trans. 22 (2000) 4052-4055. |
| [43] |
D.F. Foster, D.J. Adams, D. Gudmundsen, et al., J. Chem. Soc., Chem. Commun. 7 (2002) 722-723. |
| [44] |
M.L. Clarke, D. Ellis, K.L. Mason, et al., Dalton Trans. 7 (2005) 1294-1300. DOI:10.1039/b418193j |
| [45] |
K. Taniguchi, S. Itagaki, K. Yamaguchi, N. Mizuno, Angew. Chem. Int. Ed. 52 (2013) 8420-8423. DOI:10.1002/anie.201303132 |
| [46] |
I. Nohira, N. Chatani, ACS Catal. 11 (2021) 4644-4649. DOI:10.1021/acscatal.1c01102 |
| [47] |
J. Wang, L.E. Ehehalt, Z. Huang, et al., J. Am. Chem. Soc. 145 (2023) 9951-9958. DOI:10.1021/jacs.2c11552 |
| [48] |
J.Y. Lee, Y.H. Huang, S.Y. Liu, et al., Chem. Commun. 48 (2012) 5632-5634. DOI:10.1039/c2cc31299a |
| [49] |
J. Lee, B. Kang, D. Kim, et al., J. Am. Chem. Soc. 143 (2021) 18406-18412. DOI:10.1021/jacs.1c10138 |
| [50] |
S.Y. Hong, Y. Hwang, M. Lee, S. Chang, Acc. Chem. Res. 54 (2021) 2683-2700. DOI:10.1021/acs.accounts.1c00198 |
| [51] |
M. Lee, J. Heo, D. Kim, S. Chang, J. Am. Chem. Soc. 144 (2022) 3667-3675. DOI:10.1021/jacs.1c12934 |
| [52] |
Y. Li, Y. Dong, Y. Li, Chin. J. Org. Chem. 44 (2024) 638-643. DOI:10.6023/cjoc202307020 |
| [53] |
J.M. Medina, J.L. Mackey, N.K. Garg, et al., J. Am. Chem. Soc. 136 (2014) 15798-15805. DOI:10.1021/ja5099935 |
| [54] |
J. Shi, L. Li, Y. Li, Chem. Rev. 121 (2021) 3892-4044. DOI:10.1021/acs.chemrev.0c01011 |
| [55] |
I. Mallov, A.J. Ruddy, H. Zhu, et al., Chem. Eur. J. 23 (2017) 17692-17696. DOI:10.1002/chem.201705276 |
| [56] |
K. Kikushima, M. Grellier, M. Ohashi, et al., Angew. Chem. Int. Ed. 56 (2017) 16191-16196. DOI:10.1002/anie.201708003 |
| [57] |
C.A. Malapit, J.R. Bour, C.E. Brigham, M.S. Sanford, Nature 563 (2018) 100-104. DOI:10.1038/s41586-018-0628-7 |
| [58] |
M. Fujita, T. Hiyama, J. Org. Chem. 53 (1988) 5405-5415. DOI:10.1021/jo00258a003 |
| [59] |
F.S. Tschernuth, T. Thorwart, L. Greb, et al., Angew. Chem. Int. Ed. 60 (2021) 25799-25803. DOI:10.1002/anie.202110980 |
| [60] |
Y. Gan, G. Wang, X. Xie, et al., J. Org. Chem. 83 (2018) 14036-14048. DOI:10.1021/acs.joc.8b02498 |
| [61] |
H. Chen, S. Sun, Y.A. Liu, et al., ACS Catal. 10 (2020) 1397-1405. DOI:10.1021/acscatal.9b04586 |
| [62] |
T. Hanada, Y. Hashizume, N. Tokuhara, et al., Epilepsia 52 (2011) 1331-1340. DOI:10.1111/j.1528-1167.2011.03109.x |
| [63] |
I. Arimoto, S. Nagato, Y. Sugaya, Y. Urawa, K. Ito, H. Naka, T. Omae, A. Kayano, K. Nishiura, Eisai R & D Management Co. Ltd. US Patent Application 2007/0142640 A1, 2007.
|
| [64] |
C.J. McElhinny, F.I. Carroll, A.H. Lewin, Synthesis 44 (2012) 57-62. DOI:10.1055/s-0031-1289587 |
| [65] |
Y. Nakao, T. Oda, A.K. Sahoo, T. Hiyama, J. Organomet. Chem. 687 (2003) 570-573. DOI:10.1016/j.jorganchem.2003.08.041 |
| [66] |
L.T. Ball, G.C. Lloyd-Jones, C.A. Russell, J. Am. Chem. Soc. 136 (2014) 254-264. DOI:10.1021/ja408712e |
| [67] |
J.Y. Lee, G.C. Fu, J. Am. Chem. Soc. 125 (2003) 5616-5617. DOI:10.1021/ja0349352 |
| [68] |
K. Durka, J. Górka, P. Kurach, et al., J. Organomet. Chem. 695 (2010) 2635-2643. DOI:10.1016/j.jorganchem.2010.08.016 |
| [69] |
S.R. Wilson, L.A. Jacob, J. Org. Chem. 51 (1986) 4833-4836. DOI:10.1021/jo00375a015 |
| [70] |
W. Qu, B. Hu, J.W. Babich, et al., Nat. Commun. 11 (2020) 1736. DOI:10.1038/s41467-020-15556-7 |
| [71] |
M. Yonemoto-Kobayashi, K. Inamoto, Y. Kondo, Chem. Lett. 43 (2014) 477-479. DOI:10.1246/cl.131108 |
| [72] |
T. Hattori, Y. Suzuki, S. Miyano, Chem. Lett. 32 (2003) 454-455. DOI:10.1246/cl.2003.454 |
| [73] |
M.P. Sibi, N.E. Carpenter, e-EROS Encyclopedia of Reagents for Organic Synthesis 2001, 1–4; https://doi.org/10.1002/047084289X.rt089.
|
| [74] |
H. Maekawa, T. Nakamura, A. Yamaguchi, Electrochemistry 81 (2013) 394398. |
| [75] |
Z. Wang, S. Chang, Org. Lett. 15 (2013) 1990-1993. DOI:10.1021/ol400659p |
| [76] |
F. Yan, J.F. Bai, Y. Dong, et al., JACS Au 2 (2022) 2522-2528. DOI:10.1021/jacsau.2c00392 |