 2025, Vol. 36
2025, Vol. 36 


b College of Chemistry and Materials Science, Fujian Key Laboratory of Advanced Materials Oriented Chemical Engineering, Fujian Normal University, Fuzhou 350117, China;
c Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, Xiamen University, Xiamen 361005, China;
d State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
During the last decade, there has been an increasing interest in organo-inorganic halide perovskite solar cells (PSCs) attributed to their rapidly increasing power conversion efficiency (PCE), and the highest certified PCE achieved 26.1% [1–5]. Currently, conventional n-i-p-type devices have exhibited the highest PCEs with the lithium bis(trifluoromethane) sulfonimide salt (LiTFSI) and 4-tert-butylpyri-dine (tBP) doped spiro-OMeTAD as hole transport materials (HTM) [6–10]. However, there are still problems in this HTM that harm the device's performance and long-term stability. Firstly, the doping of LiTFSI can rescue the poor conductivity and hole mobility of the pure spiro-OMeTAD based on the interaction between TFSI- anion and spiro-OMeTAD•+, but the hygroscopic nature of LiTFSI and the migration of Li+ ions into perovskite would accelerate the decomposition of perovskite film seriously [11–16]. Secondly, since LiTFSI is easily agglomerated in HTM [17–19], tBP is used to promote the solvency of LiTFSI and reduce phase separation. But the easy evaporability of tBP due to its low boiling point leads to pinholes in the HTM, which may worsen the conductivity of HTM and the migration of metal electrode into the perovskite, and the interfacial tBP would chemically decompose perovskite films by forming a coordinated complex of [PbI2-tBP] [20–23]. Thirdly, the doping process of HTM is typically initiated by exposing the film to air for a long time (10–24 h) to wait for the slow O2 ingress into and diffusion through the HTM film, and this process is highly environment-dependent and the oxidation may not be completed [24,25].
Currently, lots of efforts have been devoted to the development of new less hygroscopic dopants to address the first problem [26,27], such as CO2 (bubble) [11], metallic salts [28–30], protic ionic liquids [31], metal-organic complexes [32–34] and ex situ synthesized spiro-OMeTAD2 •+(TFSI–)2 radicals [35,36], have also been used to replace the oxidant LiTFSI. And also, 1-dodecanethiol is introduced into LiTFSI doped spiro-OMeTAD to successfully overcome the limitations due to the hygroscopic LiTFSI and the long-time post-oxidation [24]. However, such doping usually requires the addition of volatile tBP, or other liquid additives, and the second problem is still an issue. Recently, development of new efficient HTM or even dopant-free HTMs have also come under the spotlight and the currently design strategy mainly focus on planarity, rigidity, π-conjugated extension, donor-acceptor strategy or molecular packing. Based on these strategies, for example, some molecules with carbazole cores [37], spiro cores [38,39] or other cores that enable sufficient delocalization of electrons have reached excellent performance. However, excessive pursuit of high hole mobility will often lead to the decrease of solubility of HTMs and damage of film morphology. Therefore, it is often necessary to introduce a large number of alkyl chains to solve this problem, but it will cause the disorderly molecular stacking and low charge transfer [40]. Thus, these HTMs are still limited by their inadequate performance or by the complex preparation processes [11,24,41]. In this regard, a simple method is required to simultaneously address above limitations arising from the unstable composition of the HTM for enhanced stability of HTM as well as better stability of perovskite.
Herein, we report a new doping strategy of spiro-OMeTAD by introducing disulfiram (C10H20N2S4, TETD), which is a rubber vulcanization accelerator commonly used in industry, into HTM as a low-cost additive to overcome the limitations of the classic doped spiro-OMeTAD HTM. Owing to the disulfide bond in its molecule, TETD can release reactive sulfur (after a small dose of UV light, active TETD) for rapid oxidation of spiro-OMeTAD with a more matched energy level alignment with adjacent perovskite, which greatly reduce device performance fluctuations due to the environmental dependence. In the presence of active TETD, spiro-OMeTAD radicals were stabilized by forming [spiro-OMeTAD•]+[SC(S)N(C2H5)2]−. In this situation, the free Li+ and TFSI− have the opportunity to coordinate the tBP, and the resulting favorable energy must come from the mutual interactions among the three species, the Li+, TFSI−, and tBP. As a result, the tBP volatilizing is effectively inhibited for enhanced long-term stability. Further, the TETD can simultaneously stabilize LiTFSI by suppressing its aggregation and mitigating Li+ migration through bonding Li+ with the lone pair electrons of S in C═S. At last, the introduction of TETD can also passivate the surface defects on perovskite layer because of the heteroatom. Resultantly, the optimized device exhibits a PCE of 22.94% without air oxidation, and the corresponding unencapsulate device show superior stabilities in thermal, humidity and light conditions than the device with classical doping and post-oxidation process. After 1000 h operation at the maximum power point under AM1.5G solar light (100 mW/cm2), the TETD based device still maintained 88.2% of initial PCE. This work reminds us that when lithium is not present as LixOy, free lithium ions will coordinate with tBP to fix it in HTM film, and provide a simple and universal method for high performance and HTMs with enhanced stability and low environmental dependence.
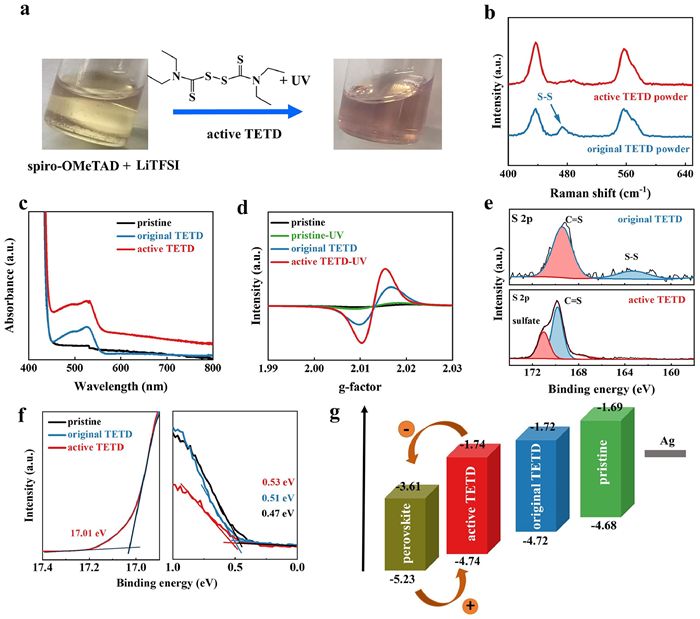
The devices with an architecture of FTO glass/TiO2 layer /Cs0.05MA0.05FA0.9PbI3 (PSK)/HTM/Ag is fabricated in this work. One-step synthesis method is used for the perovskite preparation and ethyl acetate is used for anti-solvent. The traditional spiro-OMeTAD with Li-TFSI and tBP is used as HTM in control sample, and for the TETD-included HTM, TETD solution (in chlorobenzene) was added into the pristine HTM following with a small dose of UV (365 nm) treatment to active the TETD. The effects of the introduction of active TETD on the properties of HTM and further on the device performance are studied detailed then. As shown in Fig. 1a, two changes occurred in the solution after adding active TETD into the solution. One is the color turning to pink (Fig. S1b in Supporting information) and the other is the precipitates disappearing (Fig. S1a in Supporting information). The color change shows the oxidation of spiro-OMeTAD, and the precipitate is LiTFSI due to its poor solubility in spiro-OMeTAD/chlorobenzene solution. Firstly, we explored the oxidation of spiro-OMeTAD. Considering that the S-S bond in TETD is highly active in producing free radicals, we activated the TETD with UV (356 nm, 5 mW/cm2, 60 s) to oxidize spiro-OMeTAD using the free radicals generated by TETD to further improve the carrier transport properties of HTM. Fig. 1b shows the Raman spectra of TETD, the peak at ~480 cm−1 suggests the existence of S-S bonds in original TETD, and the decrease of the peak intensity indicates the broken of disulfide bonds after UV treated [42–45].

|
Download:
|
| Fig. 1. (a) Characterization of HTM with TETD-assisted oxidation. (b) Images of spiro-OMeTAD: LiTFSI solution before and after active TETD addition. (c) Raman spectra, (d) UV–vis absorption spectra and (e) EPR signals of pristine spiro-OMeTAD, spiro-OMeTAD with original TETD or active TETD treatment. (f) XPS spectra (S 2p peak) of spiro-OMeTAD:TETD films before (original TETD) and after (active TETD) UV treatment. (g) UPS spectra of HTM films (TiO2/perovskite/HTM) and the corresponding energy levels diagram. | |
Then, to evaluate the active TETD-assisted oxidation of HTM, the ultraviolet-visible (UV–vis) was carried out on spiro-OMeTAD solution (Fig. 1c). The absorption peak at ~540 nm corresponding to the spiro-OMeTAD oxidation [46] is stronger after the addition of original TETD due to the interaction between TETD and spiro-OMeTAD, as shown in FTIR spectra (Fig. S2 in Supporting information), which may help stabling the oxidized spiro-OMeTAD+. Then, the absorption increases clearly after further UV treatment, suggesting that the generation of sulfur radicals can induce further spiro-OMeTAD oxidation. Fig. S3 (Supporting information) shows the UV–vis spectra of spiro-OMeTAD and spiro-OMeTAD:TETD with different activation methods, which identify that either heating or UV treatment can generate radicals. And the improvements of the corresponding absorbance with the increasing concentration of the active TETD as well as the increasing time of the heating or UV treatment indicate the quantitative and controllable oxidation of spiro-OMeTAD. Similarly, the electron paramagnetic resonance (EPR, Fig. 1d) of original TETD-included sample shows a radical signal, and the signal becomes more stronger after the activation of TETD by a slight UV treatment, indicating a higher concentration of spiro-OMeTAD•+ radicals in active TETD-included sample. Since the spiro-OMeTAD may also be oxidized by the UV irradiation, EPR of a pure spiro-OMeTAD solution was also tested as a control. The radical signal produced by direct UV of pure spiro-OMeTAD is much lower than that of the active TETD-based sample. XPS of S 2p for original TETD shows two peaks at 163.3 eV and 169.3 eV in Fig. 1e, which can be assigned to the S—S bonds and -C═S, respectively. After UV treated, the S—S characteristic peak of TETD disappears [47], and the peak generated at higher binding energy (~170.98 eV) indicates the formation of sulfate ions, which may be generated by the oxidation of sulfur after the S—S bond broken. The result suggests the large reactive activity of sulfur radicals. Therefore, all these results confirm the availability of the TETD-assisted oxidation. And the shifts toward lower binding energy of C1s peak in XPS spectra after the introduction of active TETD are consistent with above results on the TETD-assisted oxidation (Fig. S4 in Supporting information) [33].
The energy level of an HTM material is another important parameter that affects the device performance. Thus, to determine the effect of TETD on the energy levels of HTMs, ultraviolet photoelectron spectra (UPS) and UV–vis absorbance spectra on HTM films (film structure of TiO2/perovskite/HTM) are carried out (Fig. 1f, Fig. S5 and Table S1 in Supporting information), and the energy band diagram is shown in Fig. 1g. The addition of TETD causes the HOMO of HTM to move down (from −4.68 to −4.72 eV), and the activated TETD makes the HOMO of HTM (−4.74 eV) closer to the perovskite HOMO level (−5.23 eV), suggesting the more matched energy level alignment with adjacent perovskite.
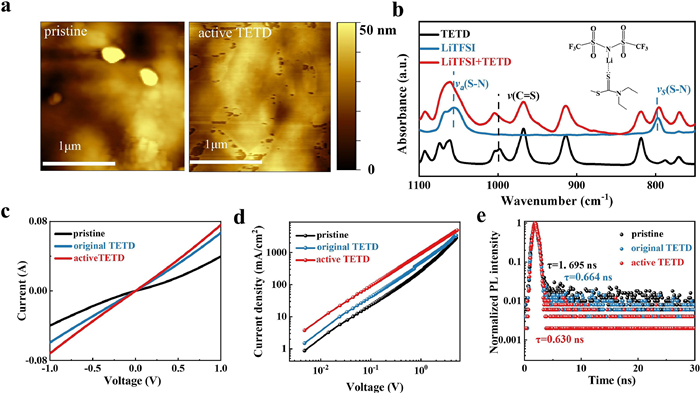
Next, we discuss another change in Fig. 1a on the precipitate disappearance. Fig. 1a and Fig. S1 (Supporting information) have shown that the LiTFSI precipitates disappeared after adding TETD into the solution, which can efficiently uniform the film morphologies, and then can promote the interfacial contact and hole transfer between the HTM and metal electrode [17,48–50]. Surface morphologies were then characterized by atomic force microscopy (AFM, Fig. 2a) and scanning electron microscopy (SEM, Fig. S6 in Supporting information) following the deposition of HTMs on the surface of the perovskite. Results indicate that the TETD-included HTM film in a smoother and more uniform manner as compared with pristine HTM, which proved by the smaller root mean square (RMS) roughness of the active TETD-included film (RMS = 4.316 nm) than the pristine HTM (RMS = 6.956 nm). In order to clarify the contribution of TETD to increasing the dissolution of LiTFSI and then improving the morphology of HTM films, the interaction between TETD and LiTFSI was studied by Fourier-transform infrared spectroscopy (FTIR, Fig. 2b). The S—N in pure LiTFSI shows a typical symmetric stretching vibration mode at 797.3 cm−1 and an asymmetric stretching vibration mode at 1055 cm−1 [24]. Because there are peaks at ~1055 cm−1 in TETD and the overlapping peaks are difficult to distinguish, thus, the symmetric stretching vibration was mainly used to identify the related interaction. The symmetric stretching vibration peak shifts to 790 cm1 when mixed with TETD. The downward shift of the S—N symmetric stretching vibration frequency may result from electron delocalization when a Lewis base–acid adduct is formed, demonstrating the interaction between TETD and LiTFSI. And the shift of C═S (~1000 cm−1) peak in the LiTFSI:TETD mixture compared with pure TETD further confirm the interaction [18,41]. Thus, the Li—S═C interaction can help improving the film morphology and enhancing device stability by suppressing LiTFSI aggregation and Li+ migration.

|
Download:
|
| Fig. 2. (a) AFM images of the surface of HTM films. (b) FTIR spectra of TETD, LiTFSI and the mixture of the two compounds. Inset in (b) is the possible interaction between LiTFSI and TETD. (c) C-V curves and (d) dark I-V of devices with the structure FTO/PEDOT:PSS/HTM/MoO3/Ag. (e) TRPL spectra of glass/PSK/HTM films. HTM is spiro-OMeTAD (with LiTFSI and tBP) w/ or w/o doped original/active TETD. | |
Then, as shown in CV (Fig. 2c) and space-charge limited current (SCLC, Fig. 2d) tests (both conducted under dark conditions with device structure of FTO/PEDOT:PSS/HTM/Ag, cm2 V−1 s−1) are superior than the pristine HTM (σ = 5.01 × 10–6 S/cm, which is consistent with the previous report [51]; carrier mobility is 9.81 × 10–4 cm2 V-1 s−1), and the parameters experience a further improvement after a slight UV irradiation (σ = 1. 21 × 10–5 S/cm, carrier mobility is 4.81 × 10–3 cm2 V–1 s–1). Clearly, the presence of TETD with UV treatment enhances the hole mobility and electrical conductivity of HTM film greatly. Further, steady-state photoluminescence (PL) and time-resolved photoluminescence (TRPL) measurements were performed to investigate the charge transfer dynamics at the perovskite/HTM interface (Fig. 2e, Fig. S7 and Table S2 in Supporting information). Results show a same enhancement trend with a short carrier lifetime of 0.630 ns for active TETD sample. Thus, we conclude that TETD helps to promote spiro-OMeTAD oxidation for enhanced hole extraction and restricted recombination in the perovskite/HTM interface without a long oxidation time [52–54].
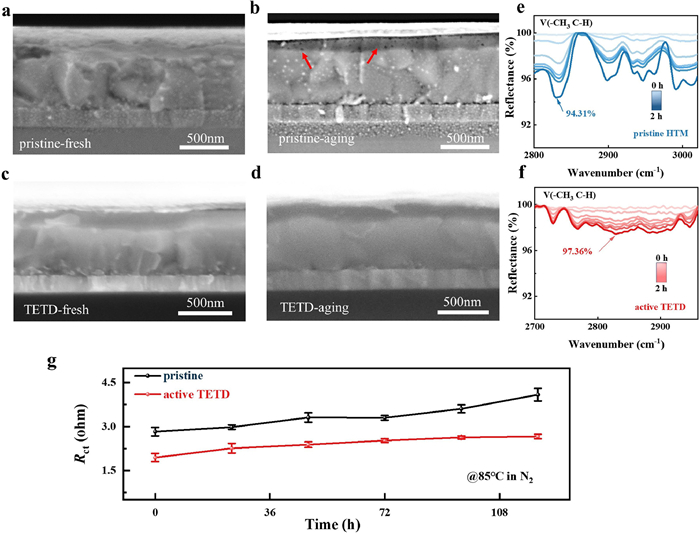
Since stability is a major challenge in perovskite devices, thus, the stability of HTM is then evaluated. In the case of pristine HTM, it is generally thought that tBP is added to increase the miscibility of the HTM solution, resulting in a better morphology, and hardly takes part in the doping reaction. However, the volatilization of tBP [55,56] and the possibility of the detrimental pyridination of spiro-MeOTAD+ remain issues in pristine HTM. As shown in Figs. 3a and b, compared with fresh devices (Fig. 3a), voids are generated in the pristine HTM film after aging (stored at 80 ℃ in air for a week, Fig. 3b) due to the escape of tBP. But in the active TETD-based HTM, problems induced by tBP seem to have been solved. Fig. 3c shows a uniform and seamless interface between perovskite and active TETD-included HTM, which could benefit the charge carrier shuttling. And the aged device with active TETD-included HTM does not show visible voids (Fig. 3d), implying that TETD can hinder tBP volatilization. Similarly, in-situ FTIR tests in a vacuum were used to monitor the concentration evolution of tBP in the HTMs. After being in a vacuum chamber, the intensity of the C—H stretching vibration peak (2800–3000 cm−1) [48,57] belonging to the pristine HTM sample gradually decreases to 94.31% after 2 h (Fig. 3e), while the corresponding peak intensity maintains at 97.36% of the original values for the TETD-included sample (Fig. 3f). Then, the evolution of charge transport properties during 120 h at a high temperature is monitored by electrochemical impedance spectrometry (EIS) measurements (Fig. 3g and Fig. S9 in Supporting information). EIS results show that the conductivity and stability of HTM film increase after introducing active TETD, even at a harsh environment of 85 ℃.

|
Download:
|
| Fig. 3. (a-d) Cross-sectional SEM images of fresh (left) and aging (right) PSC devices w/o (a, b) and w/ (c, d) active TETD. (e, f) FTIR spectra of tBP doped spiro-OMeTAD and spiro-OMeTAD:TETD stored in a vacuum chamber. (g) Rct evolution of devices during 120 h (stored at 85 ℃ in N2), the Rct values are derived from EIS spectra with a device structure of FTO/PEDOT:PSS/HTM/Ag. | |
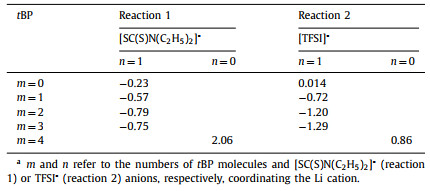
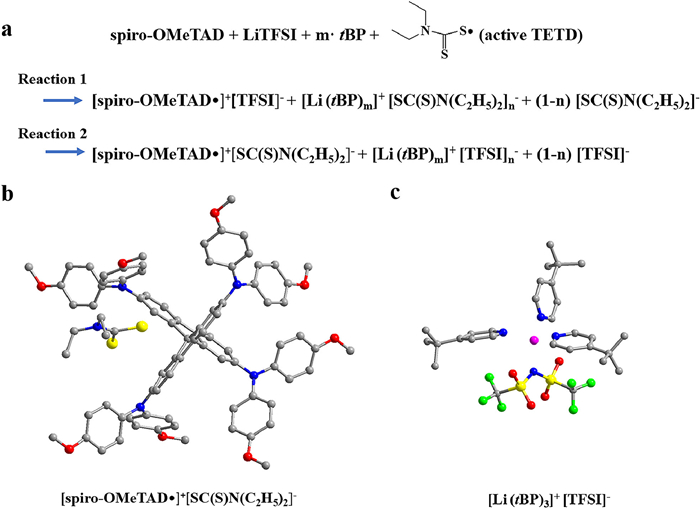
To this end, we hereby propose two possible doping mechanisms (reaction 1 and reaction 2 in Scheme 1) of the active TETD-based HTM. From the corresponding computed reaction energies (ΔE, computed by DFT calculations [58,59], Table 1), reaction 2 with m = 3, n = 1 is the most possible reaction with the lowest ΔE of −1.29 eV. Active TETD oxidizes spiro-OMeTAD, and the generated [SC(S)N(C2H5)2]- can also stabilize spiro- OMeTAD•+ free radicals by forming [spiro-OMeTAD•]+[SC(S)N(C2H5)2]-. In this situation, the free Li+ and TFSI− have the opportunity to coordinate the tBP, and the resulting favorable energy must come from the mutual interactions among the three species, the Li+, TFSI−, and tBP. Moreover, by participating in the complexation with LiTFSI, the volatile tBP molecules are expected to be ineffective and unavailable for volatilization or reaction toward the spiro-OMeTAD cation and PbI2 [41], accordingly resulting in better device stability for the case of active TETD.

|
Download:
|
| Scheme 1. (a) Reaction 1 and reaction 2 is the possible mechanism of the doping of spiro-MeOTAD with active TETD in the presence of LiTFSI and tBP. (b, c) structures of the optimized [spiro-OMeTAD•]+[SC(S)N(C2H5)2]- and [Li(tBP)3]+[TFSI]-. | |
|
|
Table 1 Computed reaction energies (ΔE in electronvolts) corresponding to the reactions in Scheme 1.a |
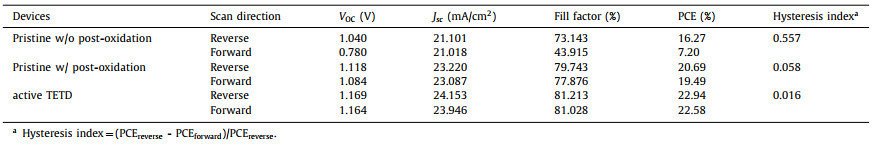
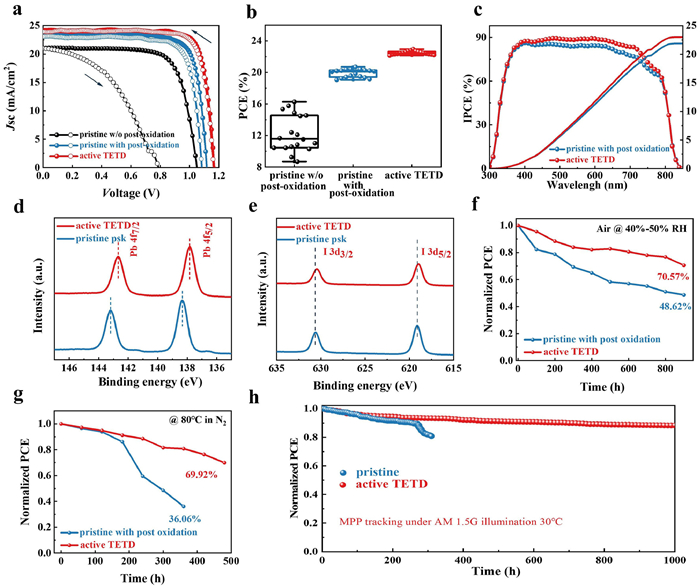
To estimate photovoltaic performance, J-V curves of corresponding PSCs were obtained (Fig. 4a, Table 2). The pristine PSCs without overnight oxidation show poor performance of 16.27% with a high hysteresis index (HI, 0.557), while the PSCs show a great improvement after overnight oxidation of HTM with an overall efficiency of 20.69%, a Jsc of 23.22 mA/cm2, a VOC of 1.118 V, a fill factor (FF) of 79.743% and a better HI of 0.058. But for active TETD-based device, it does not need to undergo a long-time oxidation to achieve a satisfied performance, and the corresponding PSCs deliver a Jsc of 24.153 mA/cm2, a VOC of 1.169 V and an FF of 81.213%, ultimately yielding a high champion efficiency of 22.94% (TETD 1 mg/mL, UV treatment time 60 s, Fig. S10a in Supporting information). The HI value also reduces to 0.016 due to the improved carrier transport and suppressed recombination in the device. The statistical distribution in Fig. 4b and Fig. S11 (Supporting information) demonstrate the reproducibility and reliability of the TETD-assisted oxidation of HTM for high performance PSCs once again. Although both the UV–vis spectra (Fig. S3b in Supporting information) and EPR (Fig. S3c in Supporting information) results shows that longer UV time can lead to higher concentration of radicals, however, as shown in Fig. S10b (Supporting information), from the point of view of device performance, the best treatment time is 60 s and longer action will degrade the performance, which may be caused by the damage of the perovskite by large dose of UV. As shown in the incident photon-to-electron conversion efficiency (IPCE) spectra, the integrated current of the target device is 23.01 mA/cm2, which is consistent with the measured JSC value (Fig. 4c). The higher Jsc and better IPCE performance of target device than the pristine HTM device shows more efficient hole extraction and transport, suggesting the efficiency of the active TETD-assistance oxidation of the HTM. As shown in Fig. S12 (Supporting information), from the external electroluminescence quantum efficiency (EQEEL) spectra, the calculated non-radiative energy loss (ΔE3) [60,61] of the active TETD-included device (0.086 eV) is lower than the pristine device with the ordinary process (0.185 eV) and is much better than that of device without a long-time oxidation (0.292 eV). These results demonstrated that the introduction of TETD could suppress the non-radiative recombination during the charge transport process, which may be caused by the reducing of damage induced by the air oxidation process, leading to an enhanced Voc.

|
Download:
|
| Fig. 4. Photovoltaic performance of PSCs with various HTM. (a) Photocurrent density-voltage (J-V) curves. (b) PCE distribution based on 18 devices. (c) IPCE and integrated Jsc of pristine and active TETD-based devices. (d) Pb 4f core level and (e) I 3d core level XPS spectra for pristine HTM and HTM with active TETD. Long-term stability of pristine and active TETD-based PSCs with 40%–50% RH in the air (f), at 80 ℃ in an N2-filled glove box (g). The original PCE of target sample are 22.45% and 22.18%, and for the control sample are 20.10% and 20.02%, respectively. (h) The stable steady-state power output of unencapsulated devices under continuous illumination using the white light-emitting diode (LED) array (100 mW/cm2) in N2 filled glove box. The original PCE is 22.41% and 20.1%, electric bias at −0.9 V. | |
|
|
Table 2 Photovoltaics parameters of devices. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Although we have used a small dose of UV light to activate TETD, the process may still cause an increase in perovskite defects because perovskite is also sensitive to UV light. Therefore, we further studied the effect of active TETD-addition on perovskite defects. As shown in Fig. S13 (Supporting information), the diode ideality factor (n) calculated from light intensity-VOC curve for active TETD-included devices is 1.1, which is lower than the pristine device (1.96), indicating the less of Shockley–Read–Hall recombination and defect in the active TETD-included samples. And in Fig. S14 (Supporting information), SCLC measurements were carried out to accurately assess the defect density of the perovskite films. As expected, the VTFL decrease (from 1.12 V to 0.89 V), leading to a decrease of the calculated defect density values (Ntrap, from 2.86 × 1016 cm−3 to 2.28 × 1016 cm−3), which is consistent with the results of diode ideality factors. Considering the S-containing group in TETD, the reduced defect density in active TETD-included samples instead of increase may be caused by the passivation effect of the TETD on the perovskite. Then, XPS spectra were further conducted to investigate the possible passivation. The shifts toward lower binding energy in Pb 4f and I 3d spectra (Figs. 4d and e) corroborate the interaction between active TETD, and the peak shift of S 2p spectra (Fig. S15 in Supporting information) of active TETD (mixed with spiro-OMeTAD) also show strong interaction of active TETD and perovskite. These XPS results further eliminate the concern of increasing perovskite defects during the HTM activation process.
Finally, the long-term stabilities of corresponding devices are scrutinized. After thermal (85 ℃ for 6 days) and humidity (50% RH for 60 days) aging tests, the X-ray diffraction (XRD) patterns of active TETD-based sample (with device structure of FTO/TiO2/perovskite/HTM) maintain a lower PbI2 (2θ of PbI2(001) = 12.7°) content than the pristine one (Fig. S16 in Supporting information), which may be caused by the perovskite passivation ability, better-blocking ability due to better morphology, suppressed Li ion migration and more hydrophobicity (due to the intrinsic hydrophobicity of TETD, Fig. S8 in Supporting information) of the active TETD-included HTM. Moreover, we also tested the long-time stability of the unencapsulated PSCs in an ambient environment (room temperature, 40%–50% relative humidity, Fig. 4f and Fig. S17 in Supporting information), continuous heating conditions (80 ℃ in N2, Fig. 4g and Fig. S18 in Supporting information) and under continuous light illumination (maximum power point tracking (MPP), 100 mW/cm2, white LED light, Fig. 4h). In an ambient environment, the active TETD-based PSCs can still maintain 70.57% of their original efficiency after 900 h of aging, while the pristine PSCs retained only 48.62% of their initial PCE under identical storage conditions. Moreover, after 500 h of heating at 80 ℃ in a nitrogen glove box, the PSCs with active TETD displayed 69.92% of their initial efficiency. In contrast, the PCEs of the pristine PSCs are only 36.06% compared to their initial efficiencies. And in MPP tracking, the optimized PSCs with active TETD can maintain 88.2% of the initial efficiency in 1000 h.
In summary, we demonstrate the effectivity of the active TETD-assistance oxidation of spiro-OMeTAD in the presence of LiTFSI and tBP. The incorporation of active TETD rapidly oxidizes the spiro-OMeTAD, penetrates tBP volatilization, and coordinates the hygroscopic LiTFSI rendering them agglomerate and migrate, which is aided to reduce the environmental dependence due to the long-time post-oxidation in air and enhance the long-term stability of PSCs. The doping mechanism is also evidenced by computational calculations. The formation of [spiro-OMeTAD•]+[SC(S)N(C2H5)2]- and [Li(tBP)m]+(TFSI-) provides a driving force with a most favorable reaction energy of −1.29 eV. The spontaneous doping mechanism under an inert atmosphere in this work provides a clear benefit from a manufacturing point of view.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementJindan Zhang: Conceptualization, Writing – original draft, Writing – review & editing, Funding acquisition, Project administration. Zhenghong Li: Data curation, Methodology, Writing – original draft, Investigation. Chi Li: Data curation, Investigation, Methodology. Mengqi Zhu: Funding acquisition, Visualization, Writing – review & editing. Shicheng Tang: Investigation, Methodology, Writing – review & editing. Kaicong Cai: Software, Writing – review & editing. Zhibin Cheng: Funding acquisition, Methodology, Writing – review & editing. Chulong Liu: Conceptualization, Funding acquisition, Investigation, Writing – review & editing. Shengchang Xiang: Funding acquisition, Project administration, Software, Writing – review & editing. Zhangjing Zhang: Funding acquisition, Project administration, Supervision, Writing – review & editing, Conceptualization.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 52001066, 21805039, 22375045, 22373015 and 22271046), the Natural Science Foundation of Fujian Province (No. 2023J01500) and Young teacher training program of Fujian Normal University (No. SDPY2023013).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110046.
| [1] |
C. Ge, P. Wang, P. Yuan, et al., Chin. Chem. Lett. 35 (2024) 109352. DOI:10.1016/j.cclet.2023.109352 |
| [2] |
Best research-cell efficiency chart, https://www.Nrel.Gov/pv/cell-efficiency.Html (accessed: July 2023).
|
| [3] |
D.L. Jia, J.X. Chen, J.M. Qiu, et al., Joule 6 (2022) 1632-1653. DOI:10.1016/j.joule.2022.05.007 |
| [4] |
A. Krishna, V. Škorjanc, M. Dankl, et al., ACS Energy Lett. 8 (2023) 3604-3613. DOI:10.1021/acsenergylett.3c01029 |
| [5] |
J.D. Zhang, S.C. Tang, M.Q. Zhu, et al., Energy Environ. Mater. (2024). DOI:10.1002/eem2.12696 |
| [6] |
J.J. Yoo, G. Seo, M.R. Chua, et al., Nature 590 (2021) 587-593. DOI:10.1038/s41586-021-03285-w |
| [7] |
M. Jeong, I.W. Choi, K. Yim, et al., Nat. Photonics 16 (2022) 119-125. DOI:10.1038/s41566-021-00931-7 |
| [8] |
B. Hu, J. Zhang, Y. Yang, et al., Chin. Chem. Lett. 35 (2024) 108933. DOI:10.1016/j.cclet.2023.108933 |
| [9] |
E.J. Juarez-Perez, M.R. Leyden, S.H. Wang, et al., Chem. Mater. 28 (2016) 5702-5709. DOI:10.1021/acs.chemmater.6b01777 |
| [10] |
G.W. Kim, H. Choi, M. Kim, et al., Adv. Energy Mater. 10 (2020) 1903403-1903432. DOI:10.1002/aenm.201903403 |
| [11] |
J. Kong, Y. Shin, J.A. Röhr, et al., Nature 594 (2021) 51-56. DOI:10.1038/s41586-021-03518-y |
| [12] |
I. Lee, J.H. Yun, H.J. Son, T.S. Kim, ACS Appl. Mater. Interfaces 9 (2017) 7029-7035. DOI:10.1021/acsami.6b14089 |
| [13] |
W.Z. Li, H.P. Dong, L.D. Wang, et al., J. Mater. Chem. A 2 (2014) 13587-13592. DOI:10.1039/C4TA01550A |
| [14] |
Z. Li, C.X. Xiao, Y. Yang, et al., Energy Environ. Sci. 10 (2017) 1234-1242. DOI:10.1039/C7EE00358G |
| [15] |
T. Wang, Y. Zhang, W.Y. Kong, et al., Science 377 (2022) 1227-1232. DOI:10.1126/science.abq6235 |
| [16] |
Y. Shen, K.M. Deng, Q.H. Chen, G. Gao, L. Li, Adv. Mater. 34 (2022) e2200978. DOI:10.1002/adma.202200978 |
| [17] |
Q. Lou, G. Lou, H.L. Guo, et al., Adv. Energy Mater. 12 (2022) 2201344. DOI:10.1002/aenm.202201344 |
| [18] |
S.G. Kim, T.H. Le, T. De Monfreid, et al., Adv. Mater. 33 (2021) e2007431. DOI:10.1002/adma.202007431 |
| [19] |
Q. Zhou, C.S. Cai, Q. Xiong, et al., Adv. Energy Mater. 12 (2022) 2201243. DOI:10.1002/aenm.202201243 |
| [20] |
S. Wang, Z.H. Huang, X.F. Wang, et al., J. Am. Chem. Soc. 140 (2018) 16720-16730. DOI:10.1021/jacs.8b09809 |
| [21] |
C. Li, J.M. Qiu, M.Q. Zhu, et al., Chem. Eng. J. 433 (2022) 133587-133594. DOI:10.1016/j.cej.2021.133587 |
| [22] |
C. Li, S.S. Guo, J.A. Chen, et al., Nanoscale Adv. 3 (2021) 3554-3562. DOI:10.1039/d1na00173f |
| [23] |
J.D. Zhang, C. Li, M.Q. Zhu, et al., Nano Energy 108 (2023) 108217-108226. DOI:10.1016/j.nanoen.2023.108217 |
| [24] |
X. Liu, B.L. Zheng, L. Shi, et al., Nat. Photonics 17 (2022) 96-105. DOI:10.3390/mca27060096 |
| [25] |
T.K. Zhang, F. Wang, H.B. Kim, et al., Science 377 (2022) 495-501. DOI:10.1126/science.abo2757 |
| [26] |
J.B. Zhang, B. Xu, L. Yang, et al., Adv. Energy Mater. 7 (2017) 1602736. DOI:10.1002/aenm.201602736 |
| [27] |
J.H. Liu, Q.S. Zhou, N.K. Thein, et al., J. Mater. Chem. A 7 (2019) 13777-13786. DOI:10.1039/c9ta02772f |
| [28] |
J. Yang, G.C. Yu, J.L. Sessler, et al., Chem 7 (2021) 3256-3291. DOI:10.1016/j.chempr.2021.10.028 |
| [29] |
J.Y. Seo, H.S. Kim, S. Akin, et al., Energy Environ. Sci. 11 (2018) 2985-2992. DOI:10.1039/c8ee01500g |
| [30] |
J.Y. Seo, S. Akin, M. Zalibera, et al., Adv. Funct. Mater. 31 (2021) 2102124-2102130. DOI:10.1002/adfm.202102124 |
| [31] |
A. Abate, D.J. Hollman, J. Teuscher, et al., J. Am. Chem. Soc. 135 (2013) 13538-13548. DOI:10.1021/ja406230f |
| [32] |
J.D. Zhang, S.S. Guo, M.Q. Zhu, et al., Chem. Eng. J. 408 (2021) 127328-127334. DOI:10.1016/j.cej.2020.127328 |
| [33] |
Y.Y. Dong, J. Zhang, Y.L. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 17610-17615. DOI:10.1002/anie.201909291 |
| [34] |
M.R. Li, D.B. Xia, Y.L. Yang, et al., Adv. Energy Mater. 8 (2017) 1702052. |
| [35] |
W.H. Nguyen, C.D. Bailie, E.L. Unger, M.D. Mcgehee, J. Am. Chem. Soc. 136 (2014) 10996-11001. DOI:10.1021/ja504539w |
| [36] |
B. Tan, S.R. Raga, A.S.R. Chesman, et al., Adv. Energy Mater. 9 (2019) 1901519. DOI:10.1002/aenm.201901519 |
| [37] |
Y.F. Wei, Y.H. Cai, L.F. He, et al., Chem. Sci. 14 (2023) 10285-10296. DOI:10.1039/d3sc02845c |
| [38] |
M. Jeong, I.W. Choi, E.M. Go, et al., Science 369 (2020) 1615-1620. DOI:10.1126/science.abb7167 |
| [39] |
J. Urieta-Mora, I. García-Benito, L.A. Illicachi, et al., Sol. RRL 5 (2021) 2100650. DOI:10.1002/solr.202100650 |
| [40] |
G. Xie, J.T. Chen, H.Y. Li, et al., Chin. J. Chem. 41 (2023) 3133-3166. DOI:10.1002/cjoc.202300234 |
| [41] |
Y. Saygili, H.S. Kim, B. Yang, et al., ACS Energy Lett. 5 (2020) 1271-1277. DOI:10.1021/acsenergylett.0c00319 |
| [42] |
B. Hernández, F. Pflüger, E. López-Tobar, et al., J. Raman Spectrosc. 45 (2014) 657-664. DOI:10.1002/jrs.4521 |
| [43] |
Y.R. Xu, D.J. Chen, Macromol. Chem. Phys. 217 (2016) 1191-1196. DOI:10.1002/macp.201600011 |
| [44] |
X. Zhang, P. Chen, Y. Zhao, M.L. Liu, Z.G. Xiao, Ind. Eng. Chem. Res. 60 (2021) 12011-12020. DOI:10.1021/acs.iecr.1c02103 |
| [45] |
J.K. Lou, L.D. Yang, T. Wei, J.M. Yuan, J.R. Deng, J. Appl. Polym. Sci. 139 (2022) e52954. DOI:10.1002/app.52954 |
| [46] |
J.X. Xia, R.L. Zhang, J.S. Luo, et al., Nano Energy 85 (2021) 106018-106029. DOI:10.1016/j.nanoen.2021.106018 |
| [47] |
W.X. Wang, Q.Q. Mao, K. Deng, et al., Small 19 (2023) e2207852. DOI:10.1002/smll.202207852 |
| [48] |
H. Yang, Y.X. Shen, R. Zhang, et al., Adv. Energy Mater. 12 (2022) 2202207. DOI:10.1002/aenm.202202207 |
| [49] |
S. Wang, M. Sina, P. Parikh, et al., Nano Lett. 16 (2016) 5594-5600. DOI:10.1021/acs.nanolett.6b02158 |
| [50] |
F.X. Cao, Z.Y. Zhu, C.H. Zhang, et al., Small 19 (2023) e2207784. DOI:10.1002/smll.202207784 |
| [51] |
J. Salunke, X. Guo, Z.H. Lin, et al., ACS Appl. Energy Mater. 2 (2019) 3021-3027. DOI:10.1021/acsaem.9b00408 |
| [52] |
X.P. Liu, B. Ding, M.Y. Han, et al., Angew. Chem. Int. Ed. 62 (2023) e202304350. DOI:10.1002/anie.202304350 |
| [53] |
X.D. Ding, H.X. Wang, C. Chen, et al., Chem. Eng. J. 410 (2021) 128328-128335. DOI:10.1016/j.cej.2020.128328 |
| [54] |
S.B. Xiong, Z.Y. Hou, W. Dong, et al., Adv. Energy Mater. 11 (2021) 2101394. DOI:10.1002/aenm.202101394 |
| [55] |
A.K. Jena, M. Ikegami, T. Miyasaka, ACS Energy Lett. 2 (2017) 1760-1761. DOI:10.1021/acsenergylett.7b00582 |
| [56] |
W.Y. Song, L. Rakocevic, R.T. Eachambadi, et al., ACS Appl. Mater. Interfaces 13 (2021) 44294-44301. DOI:10.1021/acsami.1c11227 |
| [57] |
Z. Hawash, L.K. Ono, S.R. Raga, M.V. Lee, Y. Qi, Chem. Mater. 27 (2015) 562-569. DOI:10.1021/cm504022q |
| [58] |
X.J. Mu, Y.J. Liu, G.B. Xiao, et al., Angew. Chem. Int. Ed. 62 (2023) e202307152. DOI:10.1002/anie.202307152 |
| [59] |
X.Y. Mei, J.X. Wang, X.Y. Zhang, et al., ACS Energy Lett. 8 (2023) 4386-4396. DOI:10.1021/acsenergylett.3c01686 |
| [60] |
W.J. Chen, S. Liu, Q.Q. Li, et al., Adv. Mater. 34 (2022) 2110482. DOI:10.1002/adma.202110482 |
| [61] |
Q.R. Cheng, H.Y. Chen, F. Yang, et al., Angew. Chem. Int. Ed. 61 (2022) e202210613. DOI:10.1002/anie.202210613 |