 2025, Vol. 36
2025, Vol. 36 


b University of Chinese Academy of Sciences, Beijing 100049, China;
c Fluorescence Research Group, Singapore University of Technology and Design, Singapore 487372, Singapore
Studies on the fluorescence principle have continuously progressed, culminating in the current molecular fluorescence theories that employ molecular structure to comprehend fluorescence properties [1,2]. The initial theory to comprehend such structure-property relationship of fluorophores breaks down fluorescent dyes or fluorophores into chromophore and auxochromophore components [3,4]. Subsequently, as valence bond theory progressed, it further advanced into the concept of conjugated/delocalized systems and the incorporation of electron-donating and electron-withdrawing groups [5-8]. The advent of frontier molecular orbital theory and its success in elucidating the optical properties of fluorophores placed further emphasis on the electronic structures of the fluorophores, in addition to their molecular structures [9,10]. Although our grasp of the structure-property relationship in organic fluorophores has advanced significantly, our ability to precisely tune the optical properties of these fluorophores remains constrained.
In recent years, advancements in fluorescence technology have revitalized the ancient research field of fluorescent dyes [11,12]. Innovations such as single-molecule and super-resolution fluorescence imaging [13,14], fluorescence-guided surgery [15], photodynamic [16], and photothermal [17] therapies have garnered significant attention. This resurgence in vigorous research necessitates a deeper understanding of fluorophore structures to achieve enhanced optical properties [10,18-21]. Consequently, there is an urgent need for breakthroughs in the understanding of fluorophores' structure-property relationships.
Cross-conjugation refers to a molecular arrangement in which alternating single and multiple bonds do not form a continuous conjugated system but are interrupted by non-conjugated groups or atoms. Cross conjugation in fluorophores involves the extension of conjugated systems across multiple branches or nonadjacent positions, alternating electronic communication [22]. The presence of multiple pathways for electron delocalization contributes to altered energy levels and electronic transitions, consequently influencing the absorption and emission properties [23]. However, there have been very few studies exploring cross-conjugation in fluorophores and their impact on optical properties.
In this paper, we use naphthalene fluorophores as a case study to explore the role of cross-conjugation in the optical structure-property relationship (Scheme 1). Electron-donating groups and electron-withdrawing groups are introduced at both ends of naphthalene (Scheme 1a). This arrangement induces intramolecular charge transfer, leading to the presence of the charge-separated quinone structure as one of the representative resonance hybrid structures (conjugated systems marked in blue and red, respectively, in Scheme 1a). Such fluorophores characterized by strong intramolecular charge transfer are termed charge-transfer fluorophores. When the position of the substituent changes, the conjugation pathways, and the associated cross-conjugation also change accordingly. As shown in Scheme 1b, we incorporated azetidine as an electron-donating group at one end of the naphthalene ring and introduced either a cyano group or an acetyl group as an electron-withdrawing group at different positions on the opposite end. This resulted in the formation of NaC-5Aze and NaC-4Aze, as well as NaA-5Aze and NaA-4Aze. These fluorophores are future hybridized to yield NaDC-Aze and PhDO-Aze, respectively. By theoretically analyzing the relationship between the spectral properties of NaDC-Aze and PhDO-Aze and their parent fluorophores NaC-5Aze/NaC-4Aze and NaA-5Aze/NaA-4Aze, we demonstrate that cross conjugation in fluorophores significantly influences absorption and emission properties.

|
Download:
|
| Scheme 1. Cross-conjugation in the optical structure-property relationship. (a) Cross-conjugation in charge-transfer (CT) fluorophore. (b) The structures of CT fluorophore and hybrid cross-conjugation fluorophores. | |
{kind=link}
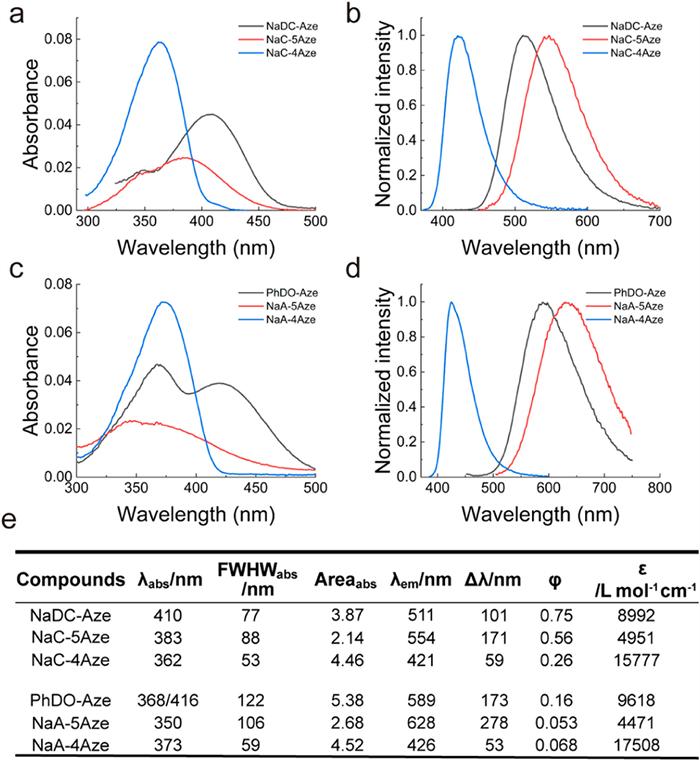
We first examined the ultraviolet–visible (UV–vis) absorption and fluorescence spectra of these 6 compounds in 10 solvents (Tables 1 and 2; Fig. S1 and Tables S1–S4 in Supporting information). For the sake of simplicity, we chose to compare their spectra in DMSO within the main text (Fig. 1). The spectra vary significantly based on the different positions of the electron-withdrawing groups. Compounds NaC-5Aze and NaA-5Aze, featuring electron-donating and electron-withdrawing groups positioned at both ends of distinct benzene rings, exhibit noticeable redshifts in their maximum absorption and fluorescence wavelengths as the solvent polarity increases. In contrast, compounds NaC-4Aze and NaA-4Aze, where the electron-donating and electron-withdrawing groups are situated at both ends of the same benzene ring, exhibit small variations in the maximum absorption and fluorescence wavelengths as a function of solvent polarity. This distinction arises from the greater intramolecular charge transfer experienced by NaC-5Aze and NaA-5Aze upon light excitation, than that in NaC-4Aze and NaA-4Aze. The significant differences in intramolecular charge transfer also lead to disparate spectral properties. For example, the molar extinction coefficients of NaC-5Aze and NaA-5Aze are only approximately one-quarter of those in NaC-4Aze and NaA-4Aze. Additionally, the first absorption bands of NaC-5Aze and NaA-5Aze are of significantly longer wavelengths than those of NaC-4Aze and NaA-4Aze. Furthermore, NaC-5Aze and NaA-5Aze exhibit large Stokes shifts (170 and 270 nm in DMSO), while NaC-4Aze and NaA-4Aze demonstrate smaller Stokes shifts (60 and 50 nm in DMSO).
|
|
Table 1 Photophysical data for NaDC-Aze in various solvents: peak UV–vis absorption wavelength (λabs), full width at half maximum (FWHMabs) and peak area (Areaabs) in UV–vis spectra, maximum emission wavelength (λem), Stokes shifts (Δλ), molar absorption coefficients (ε) and fluorescence quantum yields (φ). |
|
|
Table 2 Photophysical data for PhDO-Aze in various solvents. |

|
Download:
|
| Fig. 1. Characterization of the spectral properties of fluorophores. (a) UV–vis absorption spectra and (b) normalized fluorescence spectra of NaDC-Aze (Ex: 390 nm), NaC-5Aze (Ex: 360 nm), and NaC-4Aze (Ex: 360 nm) in DMSO. (c) UV–vis absorption spectra and (d) normalized fluorescence spectra of PhDO-Aze (Ex: 400 nm), NaA-5Aze (Ex: 350 nm), and NaA-4Aze (Ex: 370 nm) in DMSO. (e) Photophysical data of the six compounds. Dye concentrations: 5 µmol/L. | |
{kind=link}
Fascinatingly, NaDC-Aze and PhDO-Aze exhibit spectral properties reminiscent of their parent dyes. Both NaDC-Aze and PhDO-Aze display two adjacent absorption peaks, with the wavelengths aligning with the absorption peaks of their two parent dyes (Fig. 1). The molar extinction coefficient falls between the absorption intensities of the two parent dyes as well. Furthermore, the maximum emission wavelength and Stokes shift of NaDC-Aze and PhDO-Aze also fall between the maximum emission wavelengths of the two parent dyes, accompanied by higher fluorescence quantum yields compared to those of the parent dyes.
These "superimposed" spectral properties are likely the results of the hybridization within the cross-conjugation systems in NaDC-Aze and PhDO-Aze. Specifically, the presence of two absorption peaks in the absorption spectra, along with the intermediate absorption intensity, indicates the coexistence of two electron-delocalization systems in a cross-conjugation manner in both NaDC-Aze and PhDO-Aze. Upon light absorption and transition to the excited states, the delocalized system of the NaC/Nac-4Aze-type exhibits a high-energy level. Subsequently, internal conversion towards the lower-energy delocalized system of the NaC/NaA-5Aze-type results in the emission of long-wavelength fluorescence.
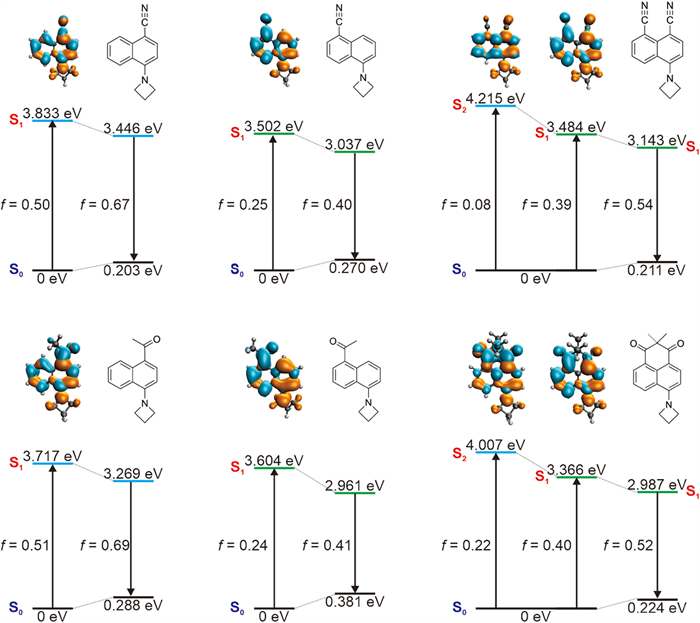
We further performed quantum chemical calculations to substantiate the experimental data (Fig. 2). Our calculations show that dye NaC-5Aze exhibits a lower optical gap (3.502 eV) compared to dye NaC-4Aze (3.833 eV), attributable to significantly enhanced intramolecular charge transfer (ICT) [24,25]. For instance, the calculated charge transfer distance, or the centroid distance between the hole and the electron, measures 2.19 Å for NaC-5Aze and 0.77 Å for NaC-4Aze. This pronounced ICT in NaC-5Aze justifies its redshift relative to NaC-4Aze. Additionally, the stronger ICT in NaC-5Aze results in a lower oscillator strength (f = 0.25), consequently leading to a reduced molar extinction coefficient. Conversely, despite its blue shifts, NaC-4Aze's reduced degree of ICT results in a higher oscillator strength (f = 0.50), explaining its large molar extinction coefficient. These findings align well with experimental observations. We also noted that the S2 state of both NaC-5Aze and NaC-4Aze show small oscillator strength, indicative of weak absorbance.

|
Download:
|
| Fig. 2. Calculated optical gaps during the UV–vis absorption and emission processes of NaC-4Aze, NaC-5Aze, NaDC-Aze (the top panel), NaA-4Aze, NaA-5Aze, and PhDO-Aze (the bottom panel). The insets show the molecular structures, oscillator strengths, and electron/hole plots during the vertical excitation of these dyes. Green: electron; orange: hole. | |
{kind=link}
Interestingly, the cross-conjugated hybrid dye NaDC-Aze exhibits an electronic and spectral structure "superimposable" on those of its parent dyes NaC-5Aze and NaC-4Aze. The S1 optical gap of NaDC-Aze (3.484 eV) is remarkably similar to that of NaC-5Aze (3. 502 eV). The charge distribution analysis reveals that in NaDC-Aze, charge predominantly migrates towards the electron-withdrawing group at the 5-position. The atomic contribution at the 5-position substituent in the lowest unoccupied molecular orbital (LUMO) of NaDC-Aze amounts to ~0.09, in contrast to the 4-position substituent which is only ~0.02. This difference mirrors the strong ICT effect observed in NaC-5Aze when an electron-withdrawing group is present at the 5-position, attracting charge density upon photoexcitation. Additionally, NaDC-Aze possesses a bright S2 state (f = 0.08) with π-π transition characteristics, leading to the manifestation of two absorption peaks within the first absorption band, corroborating experimental data.
However, the spectral characteristics of NaDC-Aze do not merely represent an additive combination of those of NaC-5Aze and NaC-4Aze. During S1 photoexcitation of NaDC-Aze, the 4-position substituents also engage in π-conjugation, although their charge density shift is less pronounced than that of the 5-position substituents. The charge transfer distance (dCT) during the S1 photoexcitation of NaDC-Aze is calculated to be 1.53 Å, resulting in a moderate oscillator strength (f = 0.39), positioning it between NaC-5Aze (f = 0.25) and NaC-4Aze (f = 0.50). Consequently, the molar extinction coefficient of NaDC-Aze at the first UV–vis absorption peak lies between those of NaC-5Aze and NaC-4Aze. Furthermore, the electron distribution in the S2 state of NaDC-Aze shows enhanced intramolecular charge transfer in comparison to the S1 state of NaC-4Aze. For instance, upon photoexcitation to the S2 state, the amino donor group in NaDC-Aze exhibits a reduced charge density compared to the charge density of NaC-4Aze's amino donor group when excited to the S1 state (Fig. S21 in Supporting information). These distinctions lead to the S2 peak absorbance in NaDC-Aze (f = 0.08) not being as pronounced as the S1 peak absorbance (f = 0.50) in NaC-4Aze.
Similar calculations performed on NaA-5Aze, NaA-4Aze, and PhDO-Aze yield analogous results. While the S1 states of NaA-5Aze and NaA-4Aze are bright, their S2 states are almost dark (f < 0.05). The cross-conjugation hybrid PhDO-Aze, however, exhibits both S1 and S2 as bright states with substantial absorbance (f > 0.20), with peak wavelengths closely resembling the S1 of NaA-5Aze and NaA-4Aze, respectively. Nevertheless, the participation of both electron-withdrawing groups in π-conjugation within PhDO-Aze, unlike in NaA-5Aze and NaA-4Aze, modifies the degree and extent of charge transfer, leading to spectral properties that echo those of NaA-5Aze and NaA-4Aze, yet are not a straightforward summation.
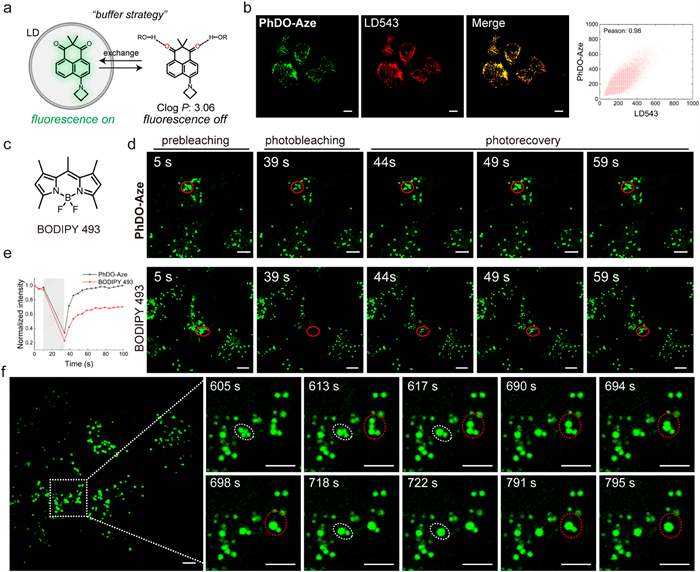
Due to the inherent environmental sensitivity of PhDO-Aze, it serves as a fluorogenic probe for wash-free imaging. Before employing the probe for imaging experiments in live cells, we initially assessed its cytotoxicity using the MTT assay. The cell viability test, conducted after incubating cells with various concentrations of PhDO-Aze for 24 h, indicated minimal impact on cell viability (Fig. S2 in Supporting information). Calculated logP (ClogP) is used to define the lipophilicity of compounds. And "ClogP < 5" is considered as strong lipophilicity for lipid droplet imaging. The calculated ClogP of PhDO-Aze is 3.06, indicating appropriate lipophilicity (Fig. 3a). Based on our prior research [14,26-28], we infer that it is an excellent buffering probe suitable for lipid droplet imaging. Through co-localization experiments, the Pearson correlation coefficient (PCC) between PhDO-Aze and LD 543, a commercial lipid droplet dye, was calculated to be 0.96 (Fig. 3b). This high PCC value indicates that PhDO-Aze has strong targeting ability to lipid droplets. Furthermore, we investigated the buffering capability of the probe through fluorescence recovery after photobleaching (FRAP) experiments. We found that PhDO-Aze rapidly recovered to its initial fluorescence intensity within 20 s post-photobleaching, while BODIPY 493, a commercial lipid droplet dye, exhibited a slower recovery rate, reaching only 60% of the initial fluorescence intensity (Figs. 3c–e). Subsequently, we employed confocal imaging to observe the dynamic behavior of lipid droplets in HepG2 cells using PhDO-Aze. Imaging results showed that multiple lipid droplets fused into one larger lipid droplet multiple times within 3 min (Fig. 3f). These results indicate that PhDO-Aze is an outstanding lipid droplet buffering probe. Owing to its substantial Stokes shift, PhDO-Aze can also serve as a powerful tool for multi-color imaging of cellular organelles, revealing additional physiological functions of lipid droplets.

|
Download:
|
| Fig. 3. Fluorogenic imaging of lipid droplets (LD) in live cells using PhDO-Aze based on the buffering strategy. (a) Mechanism for LD fluorogenic and dynamic imaging using PhDO-Aze. (b) Confocal images of HeLa cells using 1 µmol/L PhDO-Aze and 500 nmol/L LD 543. (c) The chemical structure of BODIPY 493. (d) Confocal images of live HeLa cells during photobleaching and photorecovery processes using 2 µmol/L PhDO-Aze and BODIPY 493. Red circles highlighted the bleaching area. (e) Relative intensity of the bleaching area during the photobleaching and photorecovery processes as shown in (d). (f) Confocal image of live HepG2 cells with 1 µmol/L PhDO-Aze and the locally enlarged images of the boxed region for dynamic LD coalescence. Scale bar: 5 µm. | |
{kind=link}
In this paper, we utilize naphthalene fluorophores, NaDC-Aze and PhDO-Aze, as a case study to underscore the crucial role of cross-conjugation in the structure-property relationship of fluorophores. Formed by hybridizing two distinct conjugated systems within a single naphthalene molecule, NaDC-Aze and PhDO-Aze showcase spectral characteristics from both conjugated parent systems. Both experimental data and theoretical calculations support the presence and function of two coexisting electron delocalization systems in a cross-conjugation manner within NaDC-Aze and PhDO-Aze, affording broad absorption bands, bright fluorescence, and large Stokes shifts. This work is anticipated to offer a fresh perspective on understanding the structure-property relationships of fluorophore structures and contribute to the development of novel fluorophores in the future.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementKai An: Data curation, Writing – original draft. Qinglong Qiao: Supervision. Lovelesh: Data curation. Syed Ali Abbas Abedi: Data curation. Xiaogang Liu: Data curation, Writing – review & editing. Zhaochao Xu: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 22225806, 22078314, 21908216, 22378385) and Dalian Institute of Chemical Physics (Nos. DICPI202142, DICPI202436), Agency for Science, Technology and Research (No. A*STAR, Singapore) under its Advanced Manufacturing and Engineering Program (No. A2083c0051), and SUTD Kickstarter Initiative (No. SKI 2021_03_10). The authors are grateful for the supercomputing resources of SUTD-MIT IDC and the National Supercomputing Centre (Singapore).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109786.
| [1] |
R.W. Sinkeldam, N.J. Greco, Y. Tor, Chem. Rev. 110 (2010) 2579-2619. DOI:10.1021/cr900301e |
| [2] |
E.M.S. Stennett, M.A. Ciuba, M. Levitus, Chem. Soc. Rev. 43 (2014) 1057-1075. DOI:10.1039/C3CS60211G |
| [3] |
A. Kurutos, D. Citterio, J. Mol. Struct. 1247 (2022) 131381. DOI:10.1016/j.molstruc.2021.131381 |
| [4] |
J.V. Jun, E.J. Petersson, D.M. Chenoweth, J. Am. Chem. Soc. 140 (2018) 9486-9493. DOI:10.1021/jacs.8b03738 |
| [5] |
M. Pengshung, P. Neal, T.L. Atallah, et al., Chem. Commun. 56 (2020) 6110-6113. DOI:10.1039/c9cc09671j |
| [6] |
C.X. Yan, Z.Q. Guo, W.J. Chi, et al., Nat. Commun. 12 (2021) 3869. DOI:10.1038/s41467-021-24187-5 |
| [7] |
C.S. Abeywickrama, Chem. Commun. 58 (2022) 9855-9869. DOI:10.1039/d2cc03880c |
| [8] |
X. Cheng, S. Huang, Q. Lei, et al., Chin. Chem. Lett. 33 (2022) 1861-1864. DOI:10.1016/j.cclet.2021.10.024 |
| [9] |
W. Chi, Q. Qiao, C. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 20215-20223. DOI:10.1002/anie.202010169 |
| [10] |
X.G. Liu, Q.L. Qiao, W.M. Tian, et al., J. Am. Chem. Soc. 138 (2016) 6960-6963. DOI:10.1021/jacs.6b03924 |
| [11] |
Z.G. Yang, A. Sharma, J. Qi, et al., Chem. Soc. Rev. 45 (2016) 4651-4667. DOI:10.1039/C5CS00875A |
| [12] |
C.Y. Li, G.C. Chen, Y.J. Zhang, et al., J. Am. Chem. Soc. 142 (2020) 14789-14804. DOI:10.1021/jacs.0c07022 |
| [13] |
Q.L. Qiao, W.J. Liu, J. Chen, et al., Angew. Chem. Int. Ed. 61 (2022) e202202961. DOI:10.1002/anie.202202961 |
| [14] |
J. Chen, W.J. Liu, X.N. Fang, et al., Chin. Chem. Lett. 33 (2022) 5042-5046. DOI:10.1016/j.cclet.2022.03.120 |
| [15] |
J.J. Yim, S. Harmsen, K. Flisikowski, et al., Proc. Natl. Acad. Sci. U. S. A. 118 (2020) e2008072118. |
| [16] |
X. Zhao, Q.C. Yao, S. Long, et al., J. Am. Chem. Soc. 143 (2021) 12345-12354. DOI:10.1021/jacs.1c06275 |
| [17] |
C.E. Yang, H.R. Wang, S. Yokomizo, et al., Angew. Chem. Int. Ed. 60 (2021) 13847-13852. DOI:10.1002/anie.202102640 |
| [18] |
W.J. Liu, J. Chen, Q.L. Qiao, et al., Chin. Chem. Lett. 33 (2022) 4943-4947. DOI:10.1016/j.cclet.2022.03.121 |
| [19] |
Z.F. Li, Q.L. Qiao, N. Xu, et al., Chin. Chem. Lett. 35 (2024) 108824. DOI:10.1016/j.cclet.2023.108824 |
| [20] |
J. Li, Q.L. Qiao, Y.Y. Ruan, et al., Chin. Chem. Lett. 34 (2023) 108266. DOI:10.1016/j.cclet.2023.108266 |
| [21] |
J.P. Ding, R.S. Xiao, A.Y. Bi, et al., Chin. Chem. Lett. 34 (2023) 108273. DOI:10.1016/j.cclet.2023.108273 |
| [22] |
T. Beppu, K. Tomiguchi, A. Masuhara, et al., Angew. Chem. Int. Ed. 54 (2015) 7332-7335. DOI:10.1002/anie.201502365 |
| [23] |
H. John, C. Briehn, J. Schmidt, et al., Angew. Chem. Int. Ed. 46 (2007) 449-453. DOI:10.1002/anie.200602596 |
| [24] |
X. Wu, D. Tan, Q.L. Qiao, et al., Phys. Chem. Chem. Phys. 24 (2022) 15937-15944. DOI:10.1039/d2cp00759b |
| [25] |
X.G. Liu, J.M. Cole, Z.C. Xu, J. Phys. Chem. C 121 (2017) 13274-13279. DOI:10.1021/acs.jpcc.7b04176 |
| [26] |
J. Chen, C. Wang, W.J. Liu, et al., Angew. Chem. Int. Ed. 60 (2021) 25104-25113. DOI:10.1002/anie.202111052 |
| [27] |
W. Zhou, Q.L. Qiao, Y. Tao, et al., Sens. Actuat. B: Chem. 376 (2023) 132980. DOI:10.1016/j.snb.2022.132980 |
| [28] |
N. Xu, Q.L. Qiao, J. Chen, et al., Chem. Commun. 60 (2024) 1424-1427. DOI:10.1039/d3cc05853k |