 2025, Vol. 36
2025, Vol. 36 


b Shanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, China;
c State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, China;
d Engineering Research Center of Pharmaceutical Process Chemistry, Ministry of Education, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, China;
e Shanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China
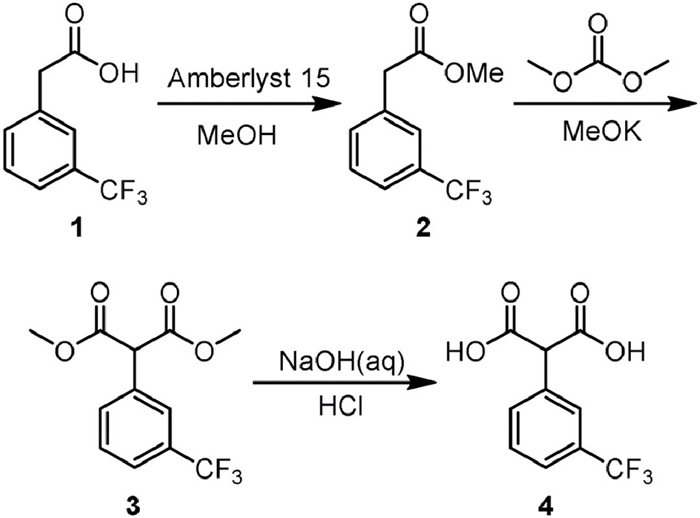
Neonicotinoids are a class of highly effective insecticides with a high market share and a wide range of applications. Recently, the large-scale application of neonicotinoid pesticides and climate anomalies have caused an acute insect resistance problem, especially in rice-related insects [1-4]. Meanwhile, the ecological safety issues of traditional neonicotinoid insecticides further limited their application [5-12]. Different from traditional neonicotinoid insecticides, Triflumezopyrim (TFM), a novel class of mesoionic pyrido[1,2-α]pyrimidinones insecticide, as inhibitor of nicotinic acetylcholine receptors (nAChRs),was developed by Dupont (Corteva) and first registered in China in 2016 [13,14]. TFM has excellent insecticidal activity against Lepidoptera and Homoptera, especially rice-related pests (Nilaparvata lugens, Sogatella furcifera, Laodelphax striatellus, etc.), and no cross-resistance with other classes of insecticides [15-19]. Moreover, TFM has low toxicity to non-target organisms, such as bees, spiders, aquatic organisms, and is eco-friendly, which makes it particularly suitable for pest control in rice-growing areas in Southeast Asia [14,15]. Several synthetic routes of 2-substituted malonic acid, the key intermediate of mesoionic pyrido[1,2-α]pyrimidinones including TFM have been reported, as shown in Fig. S1 (Supporting information). However, almost all current reported synthetic routes have the drawbacks, such as high economic cost and waste liquid, negative environmental impact (esterification), low yield and conversion (condensation), poor selectivity and prone to side effects (hydrolysis), etc. [20-37].
Compared with conventional batch mode, continuous flow synthesis has remarkable advantages in mixing performance [38], heat transfer [39], pressure control [40]. Meanwhile, on-line separation/purity technology [41], automation control [42] and modularization technology [43] in flow chemistry system greatly enhance the flexibility and compatibility of photochemistry and enzyme chemistry, etc., for large-scale green manufacture of fine chemicals [44]. Herein, for the above-mentioned shortcomings in batch mode, we firstly reported a flow chemistry approach for 2-[3-(trifluoromethyl)phenyl]malonic acid synthesis, which also offered a potential tool for synthesis and manufacture of related analogs.
Prior to conducting continuous flow studies, the designed synthetic route (Scheme 1) was initially evaluated in the batch mode with screening experiments and DoE experiments, in order to check comprehensively the influence of reaction conditions in esterification, condensation, and hydrolysis, such as equivalence ratio, solvent, reaction temperature, reaction time, reaction system concentrations. The effects were observed and recorded in Tables S1, S2 and S4, Fig. S3 (Supporting information). Subsequently, specifically optimized the parts of the process that might have a negative impact in the continuous flow synthesis.

|
Download:
|
| Scheme 1. Synthetic route of 2-[3-(trifluoromethyl)phenyl]malonic acid in this work. | |
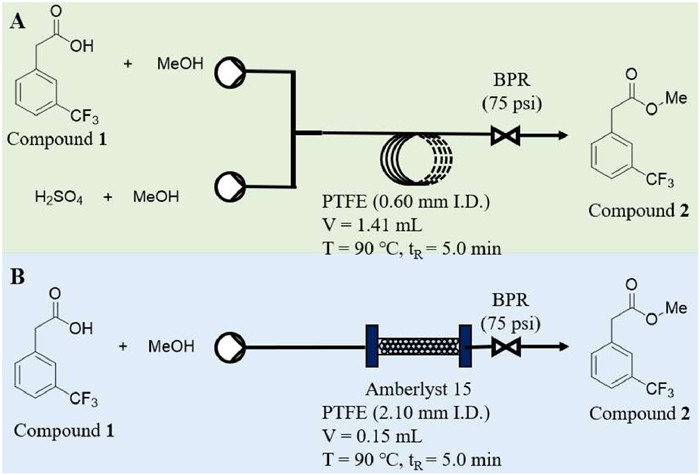
In batch mode synthesis of compound 2, sulfuric acid, p-toluenesulfonic acid, phosphotungstic acid, and Amberlyst 15 were found to have satisfactory catalytic activity and dry methanol was an optimal reaction solvent. Accordingly, two schemes were proposed to afford compound 2 based on the catalyst properties (Fig. 1). A PTFE capillary (0.6 mm i.d., 5.0 m length, shown in Fig. 1A, liquid acid as catalyst) or a stainless-steel capillary (2.10 mm i.d., 10.0 cm length, shown in Fig. 1B, solid acid as catalyst) was adopted as reactor, respectively. A back pressure regular (BPR) at 75 psi was applied at the end of the reactor coil to increase the boiling point and stabilize the flow state of the reaction system. After running a tenfold residence time, the reaction system was considered to reach a steady state, then samples were collected and analyzed in GC–MS.

|
Download:
|
| Fig. 1. Continuous flow synthesis of compound 2 from compound 1. | |
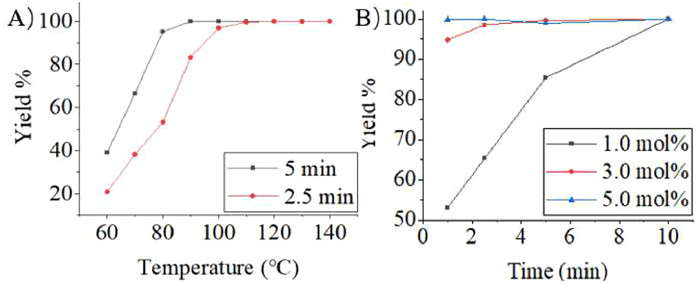
Before determining Amberlyst 15 as catalyst, the reaction system concentration of compound 1 was screened firstly under batch conditions, and the continuous mode with the same reaction conditions was used as a control. It was shown in Fig. S6 (Supporting information) that when the system concentration of compound 1 was lower than 0.5 mmol/mL, the reaction progress was slower than that in more than 1.0 mmol/mL, and reaction rate in batch mode was also dramatically slower than in continuous flow mode. It might be attributed to the fact that low substrate concentration significantly increased the distance between substrate and catalyst in reaction system, while also decreased the possibility of effective contact, so the reaction rate was slowed. Furthermore, the reaction rate of solid-liquid heterogeneous reaction system in batch mode was also particularly influenced by the solid-liquid contact area (afforded by Amberlyst 15 in this research), which was usually regarded as a key factor in heterogeneous mass transfer. Therefore, 1.0 mmol/mL system concentration of compound 1 was adopted to screen continuous process conditions. It was noteworthy that in continuous mode, the reaction effect comparable to that of 5 h in batch mode could be achieved in only 10 min, owning to the far higher mass transfer ability of micro-reactors than conventical reactors. In Fig. 2A, it was pointed out that increasing the reaction temperature promoted effectively the reaction to take place, and it only took 2.5 min and 5.0 min at 90 ℃ and 100 ℃, respectively, to achieve almost quantitative reaction. As a control, sulfuric acid was used as the catalyst instead of Amberlyst 15, and the results in Fig. 2B indicated that the reaction could be finished in 5 min with no change in other parameters, but the catalyst concentration had a significant effect on the progress of the reaction.

|
Download:
|
| Fig. 2. (A) Effect of reaction temperature with 1.0 mol% Amberlyst 15. Condition: 1.0 mmol/mL compound 1, device shown in Fig. 1B. (B) Effect of reaction time with sulfuric acid. Condition: 1.0 mmol/mL compound 1, T = 90 ℃, device shown in Fig. 1A. | |
Both Amberlyst 15 and sulfuric acid had similar catalytic effects, however, the reaction catalyzed by the former could be put into the next step directly after removing the solvent at the end of the reaction, while the latter required neutralization and extraction. Thus, Amberlyst 15 was regarded as an optimal catalyst. Considering the deactivation of Amberlyst 15 at high temperatures, and with the aim of maximize the lifetime of the catalyst, the final continuous scheme was determined to achieve 98% yield at 90 ℃ for 5 min, and the reaction time-space yield was up to 1744 g L−1 h−1.
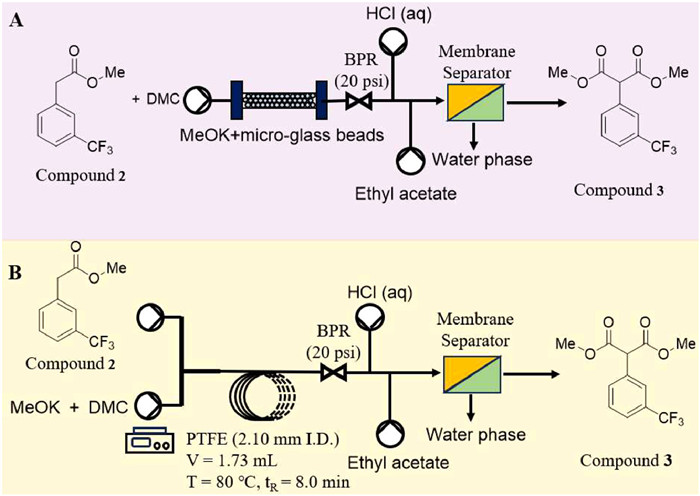
Next, potassium methanol was selected as preferred alkali for condensation to prepare compound 3 by optimizing the batch process conditions. Two continuous synthesis schemes were designed to adapt the poor solubility of potassium methanol in dry dimethyl carbonate (Fig. 3). Figs. 3A and B respectively adopted a stainless-steel capillary (10.0 mm i.d., 100.0 mm length) filled with MeOK and micro glass beads or a PTFE capillary (2.10 mm i.d., 50.0 cm length) as reactor. Membrane separator was used for on-line extraction with HCl (aq) and ethyl acetate, and flow rate was the same with reaction system respectively. A back pressure regular (BPR) at 20 psi was applied at the end of the reactor coil to control one-way flow. After running a tenfold residence time, samples were collected and analyzed in GC–MS.

|
Download:
|
| Fig. 3. Continuous flow synthesis of compound 3 from compound 2. | |
In batch mode, it was observed that the reaction system presented a significant solid-liquid heterogeneity at the initial state, then gradually transformed into a relatively stable suspension, which might be attributed to the poor solubility of potassium methanolate in dimethyl carbonate. Thus, the transportation of suspension in this reaction should be emphasized as a key in flow mode. Subsequently, two flow chemistry tactics were implemented. In Table 1, alkali and inert material were used as fillers for the packed column reactor, the reaction device was simplified in a certain degree and the average conversion rate reached 55%. It could not be ignored that in the initial stage, the reaction was almost completed, but the increase of the apparatus operation time, the reaction conversion rate decreased significantly, or even non-reaction, which was probably explained by spatial-temporal maldistribution of mass transfer in the packed bed.
|
|
Table 1 Results for continuous flow synthesis of compound 3 with device shown in Fig. 3A. |
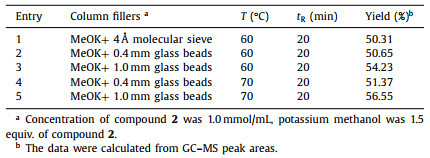
Before implementing the device shown in Fig. 3B, T-type, Y-type, and tube-in-tube mixer were tested, considering the possible influence of micro-mixers to the heterogeneous system mixing. Unfortunately, most mixers caused clogging in a short time. Although the T-mixer with shear mixing and the tube-in-tube mixer could maintain a pretty long-time stability, they still need to be flushed periodically. Hence, the tube-in-tube mixer was chosen to continuous synthesis.
In Table 2, the clogging caused by the deposition of reactants before mixing was effectively mitigated with external mass transfer intensification and the capillary diameter enlargement. As expected, high flow rates also provide some relief from clogging. It was also found that the suspension pumped while stirring was able to maintain its stability for a pretty long time. As a result, it could achieve 96% conversion in 8 min, a dramatic reduction in reaction time compared to batch reactions. Furthermore, the reaction time-space yield could reach to 882 g L−1 h−1. It was an unexpected surprise that the reaction output could be purified by extraction and distillation under reduced pressure rather than column chromatography, and the product could be used directly in the next hydrolysis reaction. Despite the problem of transporting high solids suspensions through the pipeline was solved, backflushing of the transport pipe in front of the mixer was still required at regular intervals (every 3 h) under long-term operation.
|
|
Table 2 Results for continuous flow synthesis of compound 3 with device shown in Fig. 3B.a |
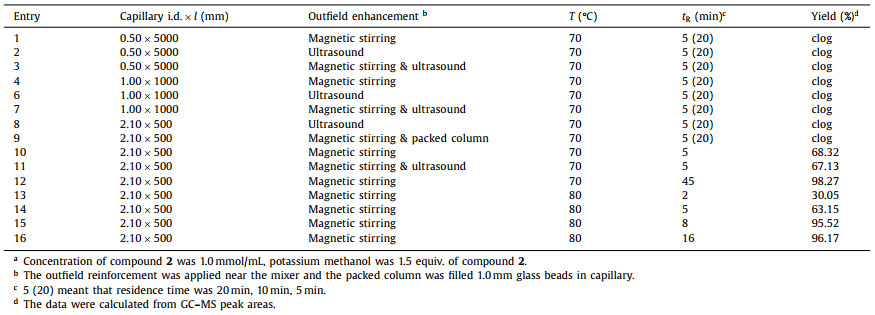
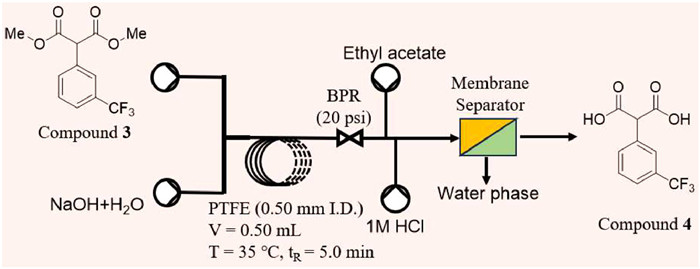
In the following hydrolysis reaction, it was found that almost all common alkali could be used in batch reaction process, and after comprehensive consideration, sodium hydroxide was finally selected for hydrolysis. A PTFE capillary (0.5 mm i.d., 2.55 m length, V = 0.50 mL) was applied as reactor. Membrane separator was used for on-line extraction with ethyl acetate and 1 mol/L HCl (aq), and flow rate was the 1.5-fold reaction system respectively. A back pressure regular (BPR) at 20 psi was used at the end of the reactor coil to control one-way flow (Fig. 4). After running a tenfold residence time, samples were collected and analyzed by HPLC.

|
Download:
|
| Fig. 4. Continuous flow synthesis of compound 4 from compound 3. | |
Due to the liquid-oil status of compound 3, the difficulty of modifying the hydrolysis reaction for continuous flow was greatly reduced. Similar to batch mode in this research, aqueous system was adopted in continuous flow mode to replace conventional water-alcohol system, which effectively decreased the consumption of chemicals and the difficulty of post-processing operations. On the other hand, in continuous flow mode, the excellent mixing and mass transfer capability of the microreactor effectively improved the defects of heterogeneous phase reaction systems observed in batch mode.
Considering that the reaction system was a liquid-liquid heterogeneous system and the comparison was about 1:10, a high-pressure mixing tree with higher mixing efficiency was chosen as the mixer in this step instead of the typical T-type mixer.
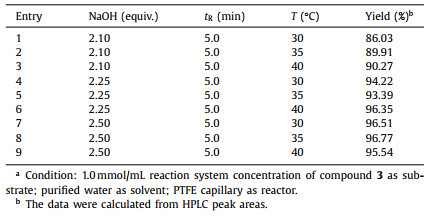
As expected, the results were essentially similar to batch mode, but the reaction efficiency was remarkably higher. As shown in Table 3 and Table S5 (Supporting information), almost all reaction conditions could achieve >97% conversion, the selectivity and yield were closely related to the base equivalent and residence time. It was manifested that both yield and selectivity at the same residence time showed positive correlation with the consumption of NaOH. A visible improvement from 2.10 equiv. to 2.25 equiv., and a subtle increasement form 2.25 equiv. to 2.50 equiv. could also be found, which might be explained by the fact that the reaction balance has been gradually approaching the end as the dosage of base increased. Besides, it was indicated that under the current conditions, the reaction could reach the end in 5 min, and increasing the temperature could shorten the reaction time but led to lower yields and selectivity.
|
|
Table 3 Results for continuous flow synthesis of compound 4 with device shown in Fig. 4.a |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
On the other hand, by analyzing the sample of the 2.5 min reaction, we found a unique peak in the liquid chromatography, which decreased markedly with the increase of reaction time and temperature until disappeared. Hence, it was presumed that it might be an intermediate in the hydrolysis process, which was a potential reason for the lower than 50% selectivity and yield of the 2.5 min reaction. After comprehensive consideration, the hydrolysis continuous process was finalized as 35 ℃, 1.0 mmol/mL dimethyl ester, 2.25 equiv. NaOH, 5 min residence time to achieve 95% conversion and 80% separation yield. In addition, the reaction time and space yield could be up to 1084 g L−1 h−1.
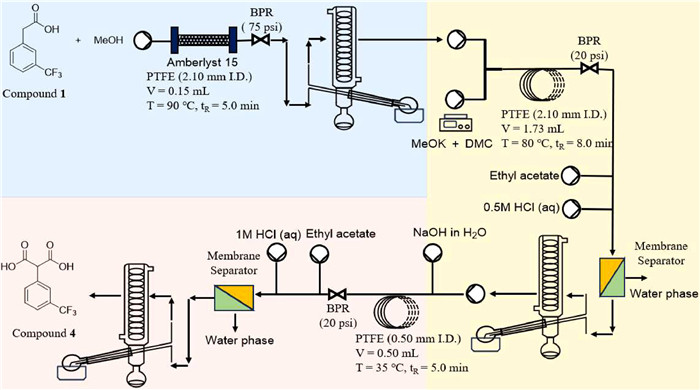
After completing the continuous flow synthesis condition optimization for each unit reaction, continuous rotation evaporation was used to connect with the above-mentioned reaction modules (Fig. 5). The reaction parameters were set to the optimal combination, and during equipment operation, samples were collected periodically at each unit output.

|
Download:
|
| Fig. 5. Semi-continuous synthesis of compound 4 from compound 1. | |
{kind=link}
Benefitting from the modification of rotary evaporator, it was possible to couple the three flow chemistry reaction units together with their corresponding post-processing units. Finally, 2-[3-(trifluoromethyl)phenyl]malonic acid (4) was prepared efficiently by three-step continuous synthesis with a total conversion of 85.64% and an overall average separation yield of about 73.38%, which was better than current reported yield (58% [26], 59% [22], 63% [27]). Moreover, the total residence time of the three-steps reaction was significantly shortened from more than 12 h to the current 18 min, which could be attributed to the excellent heat and mass transfer capabilities and exceptional mixing performance of the microreactor. Attributing to the optimization of the reaction and post-processing conditions and the application of flow chemistry, the consumption of chemical materials especially solvents and the waste generation were significantly reduced compared to batch mode while increasing reaction yields and selectivity, as shown in Table S6 (Supporting information). Considering that the post-process still has potential for optimization, the total space-time yield of the three-step reaction could be further improved from the current 98 g L−1 h−1.
In summary, we firstly reported a new flow chemistry strategy for the synthesis of 2-[3-(trifluoromethyl)phenyl] malonic acid, a key intermediate of Triflumezopyrim or mesoionic pyrido [1,2-α]pyrimidinones insecticides. Adopting inexpensive and eco-friendly reagents as starting materials, three-step reactions were achieved in three micro-reaction units. Compared with traditional batch process, continuous synthesis in all three-steps reaction owned significant advantages in terms of reaction time, reaction yield and selectivity. Finally, we achieved the synthesis of 2-[3-(trifluoromethyl)phenyl]malonic acid in 18 min with 73.38% overall separation yield. Since all the steps of the current continuous synthesis could be achieved with nearly quantitative conversion, the post-treatment was the main limitation to improve the overall separation yield. In the subsequent research, we will concentrate on the improvement of the stability of high solid-content suspensions for long time transport and online post-treatment in order to realize the long time, stable and efficient synthesis of the key intermediate of Triflumezopyrim or mesoionic pyrido[1,2-α]pyrimidinones insecticides. We deeply believed that continuous flow integration could help to achieve high-throughput screening and efficient synthesis of pesticide candidate compounds, which is of great value for the development and application of agrochemical.
Declaration of competing interestThe authors declare no competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the National Key Research and Development Program of China (Nos. 2023YFD1700303, 2022YFD17800) and National Natural Science Foundation of China (Nos. 21878088, 21476077) for financial support.
We are also grateful to Dr. Jian Zhang from Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Guizhou University, for technical communication.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109747.
| [1] |
M. Tomizawa, J.E. Casida, Annu. Rev. Pharmacol. 45 (2005) 247-268. DOI:10.1146/annurev.pharmtox.45.120403.095930 |
| [2] |
C. Bass, I. Denholm, M.S. Williamson, R. Nauen, Pestic. Biochem. Physiol. 121 (2015) 78-87. DOI:10.1016/j.pestbp.2015.04.004 |
| [3] |
J.E. Casida, Annu. Rev. Entomol. 63 (2018) 125-144. DOI:10.1146/annurev-ento-020117-043042 |
| [4] |
K. Matsuda, M. Ihara, D.B. Sattelle, Annu. Rev. Pharmacol. 60 (2020) 241-255. DOI:10.1146/annurev-pharmtox-010818-021747 |
| [5] |
A. Elbert, M. Haas, B. Springer, et al., Pest Manag. Sci. 64 (2008) 1099-1105. DOI:10.1002/ps.1616 |
| [6] |
P. Jeschke, R. Nauen, Pest Manag. Sci. 64 (2008) 1084-1098. DOI:10.1002/ps.1631 |
| [7] |
S.H. Thany, Adv. Exp. Med. Biol. 683 (2010) 75-83. DOI:10.1007/978-1-4419-6445-8_7 |
| [8] |
P. Jeschke, R. Nauen, M. Schindler, A. Elbert, J. Agric. Food Chem. 59 (2011) 2897-2908. DOI:10.1021/jf101303g |
| [9] |
M. Henry, M. Beguin, F. Requier, et al., Science 336 (2012) 348-350. DOI:10.1126/science.1215039 |
| [10] |
P.R. Whitehorn, S. O’Connor, F.L. Wackers, D. Goulson, Science 336 (2012) 351-352. DOI:10.1126/science.1215025 |
| [11] |
D. Goulson, D. Kleijn, J. Appl. Ecol. 50 (2013) 977-987. DOI:10.1111/1365-2664.12111 |
| [12] |
J.D. Crall, C.M. Switzer, R.L. Oppenheimer, et al., Science 362 (2018) 683-686. DOI:10.1126/science.aat1598 |
| [13] |
S. Du, X. Hu, M. Li, et al., Bioorg. Med. Chem. Lett. 46 (2021) 128120. DOI:10.1016/j.bmcl.2021.128120 |
| [14] |
C.W. Holyoke, W. Zhang, T.F. Pahutski, et al., ACS Symp. Ser. 1204 (2015) 365-378. DOI:10.1021/bk-2015-1204.ch026 |
| [15] |
M. Ihara, S.D. Buckingham, K. Matsuda, D.B. Sattelle, Curr. Med. Chem. 24 (2017) 2925-2934. |
| [16] |
W. Zhang, C.W. Holyoke, T.F. Pahutski, et al., Bioorg. Med. Chem. Lett. 27 (2017) 16-20. DOI:10.1016/j.bmcl.2016.11.042 |
| [17] |
D. Cordova, E.A. Benner, M.E. Schroeder, et al., Insect Biochem. Mol. Biol. 74 (2016) 32-41. DOI:10.1016/j.ibmb.2016.04.008 |
| [18] |
C.W. Holyoke, D. Cordova, W. Zhang, et al., Pest Manag. Sci. 73 (2017) 796-806. DOI:10.1002/ps.4496 |
| [19] |
H. Tan, Mod. Agrochem. 18 (2019) 42-46. |
| [20] |
C.W. Holyoke, M.T. Tong, R.A. Coats, et al., Patent, WO2009099929A1, 2009.
|
| [21] |
W. Zhang, G.D. Annis, Patent, US201161570962P, 2011.
|
| [22] |
W. Zhang, C.W. Holyoke, K.A. Hughes, et al., Patent, WO2011017342A2, 2011.
|
| [23] |
W. Zhang, Acc. Chem. Res. 50 (2017) 2381-2388. DOI:10.1021/acs.accounts.7b00311 |
| [24] |
X. Yang, Y. Ma, H. Di, et al., Adv. Synth. Catal. 363 (2021) 3201-3206. DOI:10.1002/adsc.202100369 |
| [25] |
P.T. Francis, Patent, US2013190171A1, 2013.
|
| [26] |
Y. Li, Y. Xiao, J. Lin, et al., Patent, CN112851665A, 2019.
|
| [27] |
Z. Liu, Q.X. Li, B. Song, J. Agric. Food Chem. 68 (2020) 11039-11053. DOI:10.1021/acs.jafc.0c02376 |
| [28] |
H. Guo, S. Wu, R. Song, et al., J. Agric. Food Chem. 70 (2022) 7015-7028. DOI:10.1021/acs.jafc.2c01641 |
| [29] |
T. Liu, J. Shi, D. Liu, et al., J. Agric. Food Chem. 70 (2022) 99-110. DOI:10.1021/acs.jafc.1c04715 |
| [30] |
Z. Liu, R. Song, D. Zhang, et al., Pest Manag. Sci. 78 (2022) 4983-4993. DOI:10.1002/ps.7121 |
| [31] |
J. Zhang, R. Song, S. Wu, et al., J. Agric. Food Chem. 70 (2022) 5349-5356. DOI:10.1021/acs.jafc.2c00838 |
| [32] |
D. Cai, J. Zhang, S. Wu, et al., J. Agric. Food Chem. 71 (2023) 8381-8390. DOI:10.1021/acs.jafc.3c01132 |
| [33] |
S. Hasegawa, T. Kamo, Y. Kagohara, et al., Patent, WO2016171053A1, 2016.
|
| [34] |
J. Zhang, R. Song, S. Wu, et al., J. Agric. Food Chem. 69 (2021) 15136-15144. DOI:10.1021/acs.jafc.1c05823 |
| [35] |
Z. Liu, R. Song, D. Zhang, et al., J. Agric. Food Chem. 70 (2022) 1019-1028. DOI:10.1021/acs.jafc.1c05879 |
| [36] |
D. Zhang, J. Zhang, T. Liu, et al., J. Agric. Food Chem. 70 (2022) 8598-8608. DOI:10.1021/acs.jafc.2c01899 |
| [37] |
W. Zhang, G.P. Lahm, T.F. Pahutski, K.A. Hughes, J. Agric. Food Chem. 70 (2022) 11056-11062. DOI:10.1021/acs.jafc.2c00697 |
| [38] |
M. Sun, C. Liang, L. Cao, et al., Chin. Chem. Lett. 35 (2024) 108738. DOI:10.1016/j.cclet.2023.108738 |
| [39] |
H. Jin, Q. Cai, P. Liu, et al., Chin. Chem. Lett. 35 (2024) 108721. DOI:10.1016/j.cclet.2023.108721 |
| [40] |
H. Luo, J. Ren, Y. Sun, et al., Chin. Chem. Lett. 34 (2023) 107782. DOI:10.1016/j.cclet.2022.107782 |
| [41] |
K. Wang, G. Luo, Chem. Eng. Sci. 169 (2017) 18-33. DOI:10.1016/j.ces.2016.10.025 |
| [42] |
C.P. Breen, A.M.K. Nambiar, T.F. Jamison, K.F. Jensen, Trends Chem. 3 (2021) 373-386. DOI:10.1016/j.trechm.2021.02.005 |
| [43] |
P. Liu, F. Zhao, J. Zhang, et al., Chin. Chem. Lett. 35 (2024) 109020. DOI:10.1016/j.cclet.2023.109020 |
| [44] |
B. Gutmann, D. Cantillo, C.O. Kappe, Angew. Chem. Int. Ed. 54 (2015) 6688-6728. DOI:10.1002/anie.201409318 |