 2025, Vol. 36
2025, Vol. 36 


b Key Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan Characteristic Plant Extraction Laboratory, Yunnan Key Laboratory of Research and Development for Natural Products; School of Pharmacy and School of Chemical Science and Technology, State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Yunnan University, Kunming 650091, China;
c School of Pharmacy, Nantong University, Nantong 226001, China;
d School of Pharmaceutical Sciences, Jilin University, Changchun 130021, China;
e State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China;
f College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, China;
g Faculty of Pharmaceutical Sciences, Toho University, Funabashi, Chiba 274-8510, Japan;
h School of Life Science and Engineering, Southwest Jiaotong University, Chengdu 610031, China
In recent decades, drug discovery has made significant progress, with natural products (NPs) and their derivatives playing an irreplaceable role as important sources for finding new biologically active molecules and scaffolds in medicinal chemistry [1]. NPs serve as crucial sources for new drugs and are essential for treating human diseases, owing to their rich chemical diversity and biological activities, which offer valuable resources and potential for drug development [2]. Diterpenoids can be produced by plants, microorganisms, and animals in nature, with rich structural skeletons. Their biological activities, such as anti-tumor, antibacterial, antiviral, anti-inflammatory, and analgesic activities, are remarkable in the conception of new drugs [3-8]. For example, paclitaxel, a taxane-type diterpenoid, is the renowned natural-source cancer remedy and mainstay in treating breast, lung, and ovarian cancer. Andrographolide, a labdane-type diterpenoid, has been verified to be effective against upper respiratory tract infection and bacillary dysentery in several clinical dosage forms in China. Oridonin is an ent‑kaurane type tetracyclic diterpenoid, displaying a broad range of biological effects including anticancer activities, neuroprotective, anti-inflammatory, etc. Natural diterpenoids represent a rich and untapped resource in drug discovery, owing to their structural complexity and pharmacological versatility.
With advancements in isolation, characterization, and synthesis techniques have uncovered more novel diterpenoids [9-12]. Their diverse chemical structures and pharmacological activities make them attractive candidates for therapeutic development. This review provides a comprehensive overview of the current state of research on representative natural diterpenoids, highlighting their structural diversity, pharmacological properties, and potential applications in drug discovery. Through elucidating their biological activities and mechanisms of action, this paper aims to underscore the importance of natural diterpenoids as a valuable source of novel therapeutics.
1. Chain and monocyclic diterpenoidsYingjie Wang, Ning Li*
1.1. Chain diterpenoidsChain diterpenoids, which are also known as acyclic or linear diterpenoids, originate from geranylgeraniol, a C20 metabolite with four double bonds and five methyl functions [13,14]. Their multiple positional replacements and rearrangements, such as oxidation, isomerization, unsaturated or terminal ring systems (like furan rings) formation, and polymerization and degradation modes, produce a diverse selection of derivatives with both structural and biological significance [6,15]. Chain diterpenoids is not common in nature.
In the last decade, there have been only 72 new acyclic diterpenoids reported, and their detailed information, including trivial names, structures, sources, and bioactivities, is shown in Table S1 (Supporting information). The abundance of acyclic diterpenoids have been found in recent years mainly in the genus Aphanamixis of Meliaceae family [17-22], the genera Siegesbeckia and Carpesium of Asteraceae family [22,23], the genus Croton and Vernicia of Euphorbiaceae family [24,25], and the genus Bifurcaria of brown algae [26-29].
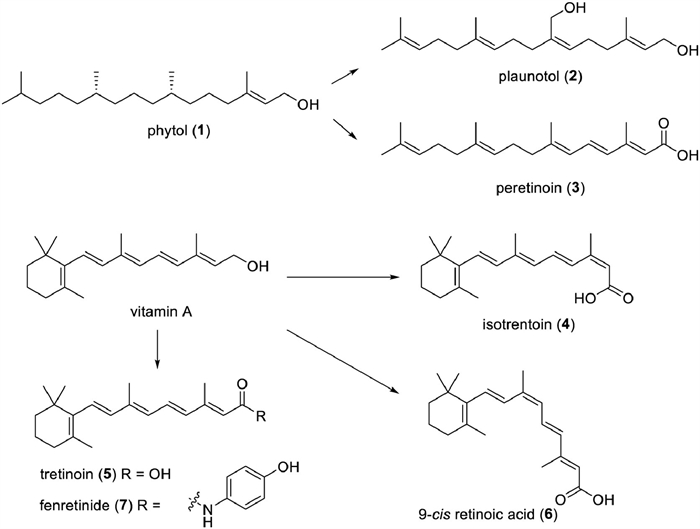
Phytol (PYT, 1) is a long-chain unsaturated diterpene, found abundantly in nature as a component of chlorophyll (Fig. 1). PYT could be considered a promising drug candidate. Several recent studies have highlighted the diverse biological effects of PYT, including antimicrobial, cytotoxic, antitumoral, antioxidant, antinociceptive, anti-inflammatory, anxiolytic, metabolism-modifying, autophagy- and apoptosis-inducing, immune-controlling, and antimicrobial effects [30,31].

|
Download:
|
| Fig. 1. The structures of drugs or drug candidates from chain and monocyclic diterpenoids. | |
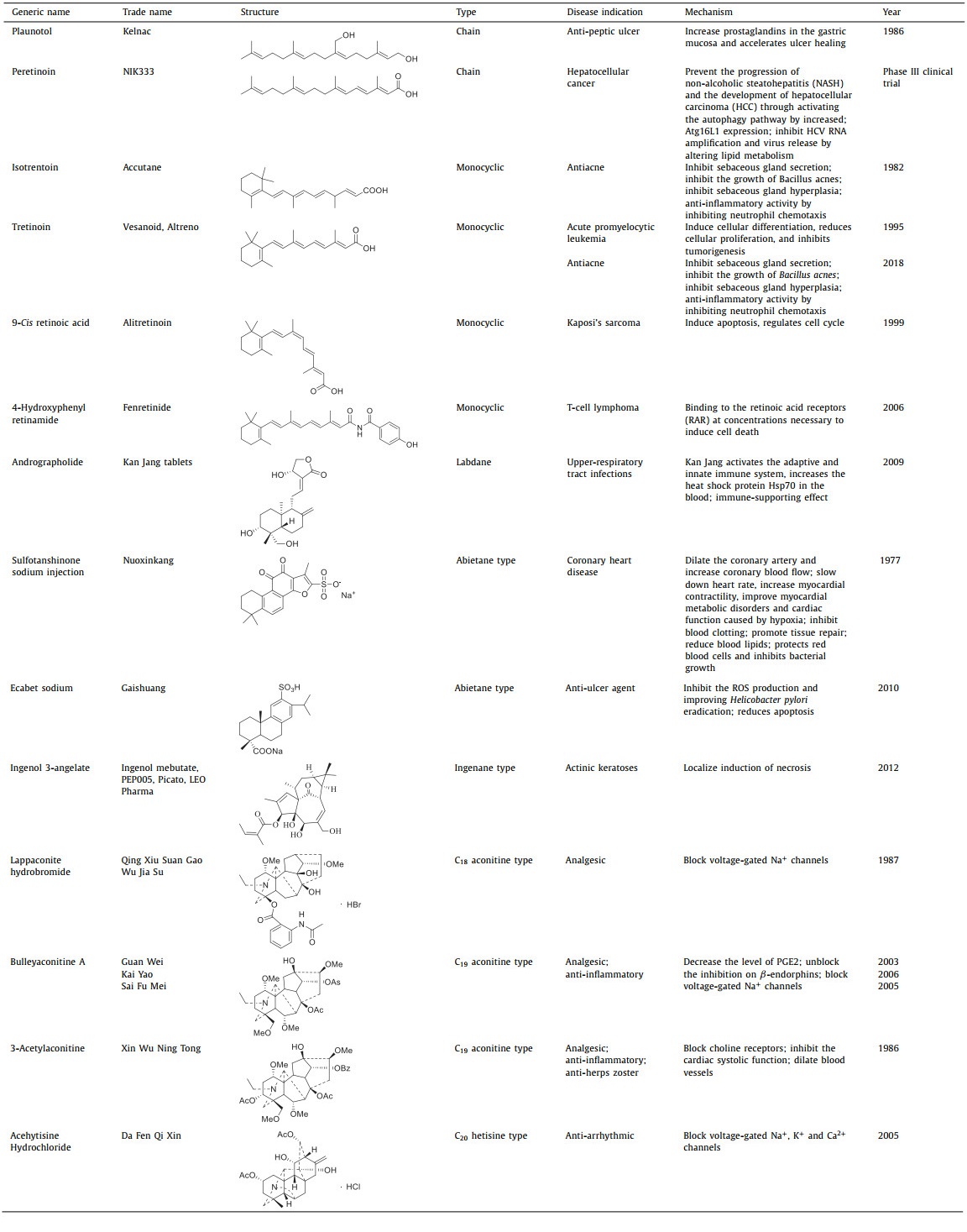
Plaunotol (KelnacTM, 2), an orally active acyclic diterpene alcohol, was first isolated from branches and leaves of Croton sulblyratus [23], showing an anti-reserpine activity [15]. In 1986, it was approved to be an anti-peptic ulcer agent in Japan and was manufactured in the form of a soft gelatin capsule. Recent pharmacological studies revealed that Plaunotol has anti-inflammatory effects [23,32,33]. Plaunotol is also able to inhibit tumor angiogenesis and cell proliferation, obstruct adriamycin-induced kidney cell death, and strengthen melanoma cells to drug-induced cell death. However, it has no significant protective effect on human lung carcinoma cells [34].
Peretinoin (NIK333, 3) has been proposed as a possible chemotherapeutic agent for hepatocellular cancer. It has been specifically formulated for secondary chemoprevention of those who had gone through curative surgery or local ablative treatment for hepatocellular carcinoma (HCC), and a Phase Ⅲ clinical trial is currently in progress [35-37]. It is also employed as a complementing therapy for those with hepatitis C, hepatitis B virus-related HCC, and nonalcoholic steatohepatitis (NASH) [38,39]. Additionally, peretinoin therapy by oral administrationbrings about an improvement of the sensorimotor function and a diminishment of the pathological changes in the striatum associated with intracerebral hemorrhage (ICH) in mice, potentially by impeding microglial nuclear factor kappa-B (NF-κB) pathways [40].
1.2. Monocyclic diterpenoidsMonocyclic diterpenoids, with a ring structure consisting only of carbon atoms, are formed by combining acyclic diterpenoids. Vitamin A and its derivatives, which are mainly derived from animal livers, are the most well-known monocyclic diterpenes. However, they are scarce in plants, only nine new monocyclic diterpenoids have been reported in the last decade (Table S2 in Supporting information). To date, monocyclic diterpenoids are only found in a few plants of Asteraceae, Podocarpaceae, Saururaceae, and Euphorbiaceae family [25,41-43], as well as in marine sponges [44].
Vitamin A and its derivatives have links to a range of physiological functions. Some of these compounds are commonly employed in food, nutrition, cosmetics, and others. Numerous naturally occurring bioactive molecules are converted into medications. For example, isotrentoin (13-cis-RA, 4), a physiologically active metabolite of vitamin A (Fig. 1), is mainly derived from animal liver and cod liver oil. It was first synthesized in 1955 [45]. The U.S. Food and Drug Administration (FDA) gave it the go-ahead for the clinical treatment of cystic acne in 1982 under the trade name AccutaneTM. Presently, oral preparations (isotrentoin soft capsule, capsule, gels) are used clinically to treat severe acne, particularly nodular cystic acne, and even for hair pityriasis and other diseases. Moreover, external use is an option for treating mild acne [46-48]. Nonetheless, a few studies have suggested that it could have some side effects, thus it should only be used following medical advice [49-51]. In addition, evidence has demonstrated that isotretinoic acid can manage the Toll-like receptor-2, and carry out immunomodulatory [52,53].
Tretinoin (all-trans-retinoic acid, ATRA, 5) is the drug of choice for treating acne and acute promyelocytic leukaemia (APL). It was approved in 1995 under the brand name Vesanoid for the treatment of APL. In 2018, the FDA approved Altreno (tretinoin, Retinoic acid Emulsion 0.05%) for use on the skin to treat acne vulgaris in those 9 and older, becoming the first retinoic acid product in emulsion form. Additionally, pharmacological research has demonstrated that ATRA has the following activities: antiproliferative [54], anticancer [55-59], anti-diabetic [60], antiviral [61,62], antibacterial [63], anti-arteriosclerotic [59,64], anti-inflammatory [65-67], and hepatoprotective activities [68]. ATRA can also be applied as an auxiliary therapy for multiple cancers, such as breast cancer and non-small cell lung cancer, minimizing adverse effects and/or increasing the potency of drug therapy.
9-Cis retinoic acid (Alitretinoin, 9-cis RA, 6), also called alitretinoin, has been utilized clinically since 1999 as a treatment for Kaposi’s sarcoma [69,70]. It has been proven to induce apoptosis, regulate cell cycle, and provide anticancer, anti-inflammatory, and neuroprotection benefits [71-76].
Fenretinide (7), belonging to the third-generation vitamins, was put into use in 2006 for treating T-cell lymphoma [77]. And it impedes the growth of many kinds of tumor cells, including neuroblastoma cells [78,79], small cell lung cancer cells [80], and breast cancer cells [81]. Fenretinide also improves intestinal barrier function and mitigates alcohol liver disease [82].
A chem-stamp process was devised to construct isoprene units from readily available aldehydes and 2,3-allenols in an effective way, which allowed for the organized and concise synthesis of many natural and pharmaceutically relevant diterpenoids (including vitamin A, retinoic acid, fenoretinoic acid, avitoic acid, peretinoin). By the abundant availability of aldehydes and 2,3-allenols as starting materials, a new idea for the source of terpenoids is furnished [83].
Therefore, monocyclic diterpenoids and retinols, with their rich biological activities, are promising candidates for the development of new drugs.
1.3. AcknowledgmentThis work was financially supported by the grant from the National Natural Science Foundation of China (No. 82373764).
2. Labdane-type and clerodane-type diterpenoidsPeng Tang, Wenchao Tu, Weilie Xiao*
As a crucial class of diterpenoid compounds, bicyclic diterpenoids derived their name from the bicyclic structure integral to their molecular structure. The molecular structures of these compounds are profoundly diverse and complex, and more than 10,000 distinct bicyclic diterpenoids have been discovered. These compounds exhibit a range of biological activities, including cytotoxicity, anti-inflammatory, antioxidant, immune regulation, antidepressant, spasmolytic, antibacterial, and numerous other significant bioactivities [84-86]. However, the potency and impact of these bioactivities vary depending on the specific bicyclic diterpenoid, thus influencing their potential for clinical utilization.
With the continuous improvement of separation technology, many trace bicyclic diterpenoids have been successfully isolated and identified, which not only enriches the skeleton types and compounds quantity of bicyclic diterpenoids but also greatly enriches the library of candidate compounds for active drug screening. This section provides a summary of the types of bicyclic diterpenoids that have high bioactivity and potential for pharmaceutical use (Fig. 2). It also covers the structure-activity relationships (SARs) of these highly bioactive bicyclic diterpenoids, as well as their potential for drug modification as lead compounds. By exploring the value of bicyclic diterpenoids in the field of bioactivity discovery and new drug development, we can gain a better understanding of their potential as valuable resources for future pharmaceutical research.

|
Download:
|
| Fig. 2. The structure of part of high bioactive bicyclic diterpenoids. | |
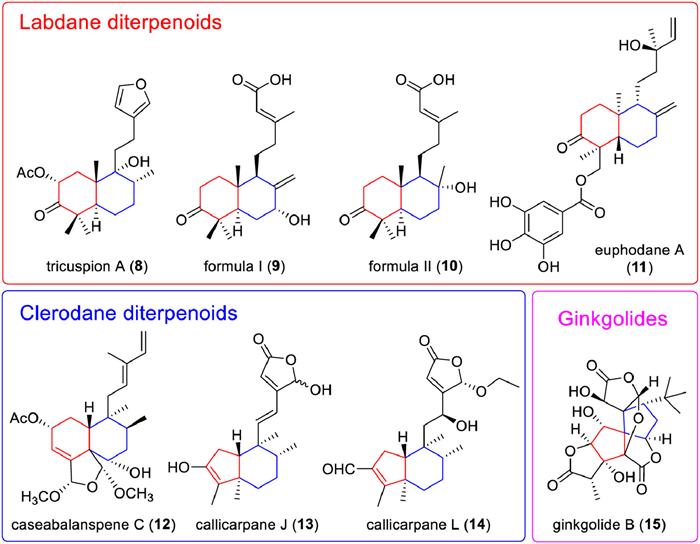
Labdane diterpenoids are a class of bicyclic diterpenoids with decalin as the parent nucleus. Diterpenoids such as formononetin, andrographolide, and phyllodin E are all bicyclic diterpenoids with this skeleton. Labdane diterpenoids are widely distributed in plant species, with diverse species and various bioactivities. Up to 7000 labdane diterpenoids have been discovered, many of which exhibit strong anti-inflammatory, anti-microbial, anti-mutagenic, and cytotoxic bioactivity [87]. In the last 10 years, a series of new compounds (Table S3 in Supporting information) with excellent bioactivity were extracted. For example, tricuspion A (8) extracted from Salvia tricuspis Franch exhibited potent anti-inflammatory activity with half maximal inhibitory concentration (IC50) value of 14.92 ± 0.51 µmol/L in lipopolysaccharide (LPS)-stimulated BV-2 microglia cells [88]; two new labdane diterpenoids (formula Ⅰ and Ⅱ, 9 and 10) isolated from the leaves of Callicarpa nudiflora exhibited potential anti-inflammatory activity with IC50 values of 10.3 ± 0.3 µmol/L and 12.7 ± 0.4 µmol/L, respectively [89]. Besides, euphodane A (11) isolated from Euphorbia pekinensis exhibited strong antitumor effects, with an IC50 of 5.92 µmol/L against U-937 cell lines [90].
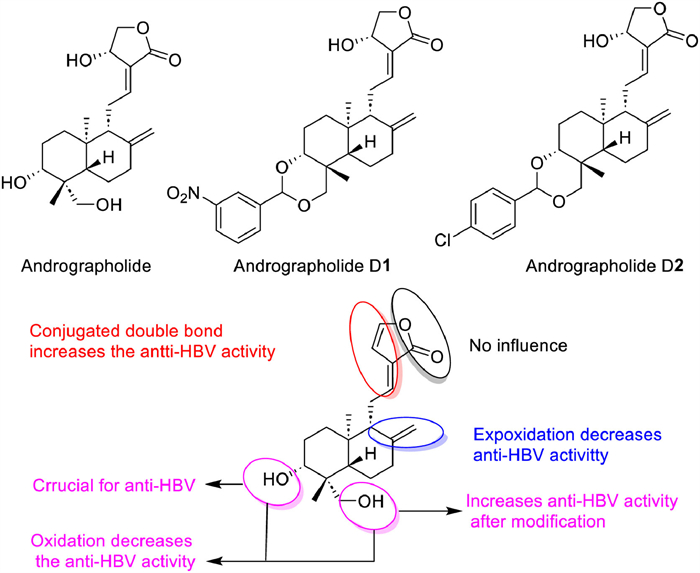
As the typical compound of labdane diterpenoids, andrographolide showed versatile bioactivities including anti-inflammation, anti-cancer, anti-obesity, anti-diabetes, anti-malaria, antioxidant, antihypertensive, antibacterial, antisickling, antiviral, and other activities [90-93]. And now, some Chinese patent drug containing andrographolide have been put into clinical, such as Kan Jang tablets, andrographolide tablets, andrographolide drop-pills, and andrographolide capsules (Table 1) [94,95]. In addition, some clinical trials for andrographolide in the prevention and treatment of related diseases are in full swing, which will great promote the clinical transformation of andrographolide products [96]. However, compared with other clinical drugs, the therapeutic effect of andrographolide is not satisfied, but its unique bicyclic skeleton structure and strong bioactivity make it one of the most promising lead compounds for molecular modification [97]. To increase bioavailability and functionality, chemical modifications on andrographolide are often performed on the α,β-unsaturated γ-butyrolactone moiety, the two double bonds C-8/17 and C-12/13, and three hydroxyls at C-3, C-14 and C-19, and a comprehensive SAR on andrographolide was achieved (Fig. 3). Among them, nitro-benzylidene derivatization andrographolide compounds D1 and D2 have shown extremely high anti-human immunodeficiency virus (HIV) activity (IC50 = 0.51 µmol/L) and strong anti-colon cancer effects (the concentration that causes 50% growth inhibition, GI50 = 0.626 µmol/L) [98]. Currently, the therapeutic evaluation of andrographolide and its derivatives for different inflammatory diseases has entered the clinical trial stage. In addition, new formulations based on drug delivery systems are constantly being developed to improve the therapeutic coefficient and druggability of andrographolide molecules by improving the solubility, targeting, and pharmacokinetic properties of andrographolide and its derivatives [99,100].
|
|
Table 1 Marketable and clinically used diterpenoids drugs. |
{kind=link}
{kind=link}

|
Download:
|
| Fig. 3. Andrographolide and representative derivatives as well as its SAR. | |
{kind=link}
Clerodane diterpenoids are an intriguing subset of natural compounds synthesized from geranyl pyrophosphate as a precursor compound in plants (Table S4 in Supporting information). The basic skeleton of clerodane diterpenoids could be divided into two fragments: a fused ring decalin moiety (C-1-C-10) and a six-carbon side chain at C-9 (C-11-C-16). The remaining four carbons (C-17-C-20) are attached at the fused ring decalin [84]. Clerodane diterpenoids are highly relevant to anti-inflammation, and ent‑clerodane diterpenoids exhibit better anti-inflammatory activity than clerodane type, and the lactone rings at C-18 and C-19 are crucial for maintaining the anti-inflammatory activity of clerodane diterpenoids [101]. Till now, more and more new clerodane diterpenoids have been discovered and showed potential anti-inflammation effects. For example, caseabalanspene C (12) containing an N-heterocycle extracted from Casearia velutina showed inhibitory activity for NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome activation with IC50 of 2.90 µmol/L against lactate dehydrogenase (LDH) release [102]. Callicarpanes J (13) and L (14) extracted from Callicarpa integerrima showed significant inhibition for NLRP3 inflammasome activation with IC50 of 0.16 ± 0.24 µmol/L and 0.25 ± 0.18 µmol/L against LDH release, respectively [103]. Furthermore, a total of 6/6 ent‑clerodane diterpenoids, including 52 new compounds, were isolated from Callicarpa arborea, and 10 of them showed potent inhibitory activity against pyroptosis with IC50 < 5 µmol/L in our group. A potential SAR of clerodane diterpenoids in Callicarpa arborea on pyroptosis inhibition was established, which further clarified the anti-inflammatory activity trends of various clerodane diterpenoids in Callicarpa arborea (Fig. 4) [104]. Moreover, clerodane diterpenoids also perform other bioactivities including sedation, antidepressant, antipruritic, antitumor [87], antiaddictive, antiplasmodial [105], immunosuppression, antiosteoporosis [106], antibacterial [107]. Till now, though lots of new clerodane diterpenoids have been discovered, the clinical application of these compounds as anti-inflammatory or other therapies is still far away.

|
Download:
|
| Fig. 4. Potential structure-pyroptosis inhibition relationship of clerodane diterpenoids in Callicarpa arborea. | |
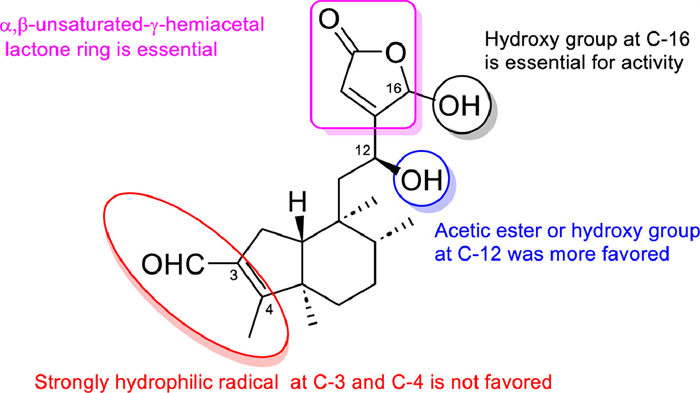{kind=link}
As the typical and advanced clerodane diterpenoid, salvinorin A is the first naturally occurring non-nitrogenous compound-type κ-opioid receptor selective agonist and non-alkaloidal hallucinogen and was considered a safer class of anesthetic and psychiatric drugs [108]. Salvinorin A has been a lead compound for the discovery of other more effective drug candidates and the SARs of salvinorin A have focused on several main areas: (1) the acetoxy group at C-2, (2) the carbomethoxy group at C-4, (3) the carbonyl at C-17, and (4) the furan ring [109].
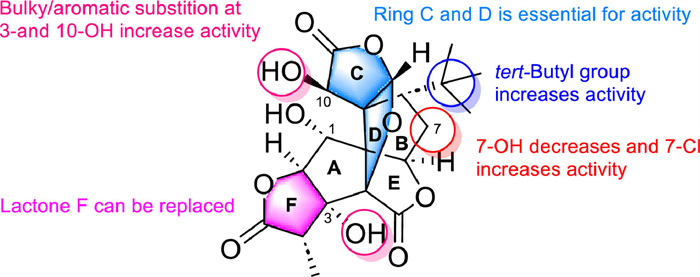
2.3. Specific bicyclic diterpenoidsGinkgolides are a specific class of bicyclic diterpenoids found only in ginkgo biloba. The skeleton of ginkgolides contains two carbon rings (a spiroadamantane ring and a tetrahydrofuran ring), and three lactone rings. Currently, seven ginkgolides have been discovered and are recognized as antagonists of platelet-activating factor receptors (PAFR). The most active against PAFR is ginkgolide B (GB, 15).
The SAR of GB against PAFR shows the lactone rings C and D as well as the 8‑tert‑butyl group are necessary for maintaining its anti-PAFR activity, while the replacement of 1-OH and 10-OH with large or aromatic groups can enhance its anti-PAFR activity. The presence of a hydroxyl group on 7-C could reduce its activity, but once another element is introduced on C-l, the anti-PAFR ability is increased (Fig. 5). Detailed SAR makes GB work as an ideal lead compound for the drug design of blood circulation disorder modulators [110]. Some ginkgolides or their derivatives have been put into clinical use or are in the clinical trial stage [111].

|
Download:
|
| Fig. 5. The SAR of GB against PAFR. | |
{kind=link}
At present, with the continuous improvement of modern separation technology and the complete coverage of the separation range of plant secondary metabolites, more and more new bicyclic diterpenoids are being continuously discovered, enriching the diversity of this type of compounds, many of which exhibit excellent bioactivity and are important sources of new drug discovery or drug modification.
On the other hand, due to the natural properties of “multiple targets, more bioactivities” in natural products, most bicyclic diterpenoids have only verified part of their bioactivities, and the utilization of the bicyclic diterpenoid is limited [112]. Structure-activity relationships can reflect the bioactivity trends of a certain type of structurally similar compounds [113]. Based on the known bioactivity data on existing bicyclic diterpenoids, it is possible and necessary to construct a database of SARs for different types of bicyclic diterpenoids with different bioactivities. This not only enables the retrieval of highly active compounds of existing bicyclic diterpenoids for different activities but also provides big data support for analyzing the biological activities of newly discovered bicyclic diterpenoids to screen out their potential pharmaceutical value.
2.4. AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 82260682, 81860615, 21762048, 81903541, 22167021 and 82260694), Project of Yunnan Characteristic Plant Screening and R&D Service CXO Platform (No. 2022YKZY001), the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT_17R94), Yun Ling Scholar Project to Wei-Lie Xiao, and the Postgraduate Research and Innovation Foundation of Yunnan University (No. KC-22222024).
3. Abietane-type diterpenoidsQi Gao, Wenli Wang*
Abietane-type diterpenoids are biosynthesized via the mevalonic acid pathway by condensation of four isoprene units (C5H8). The basic abietane core skeleton is composed of a tricyclic perhydrophenanthrene of normal series, suggesting that the methyl group (C20) attached to carbon C10 is always assigned β-oriention, two methyl groups (C18 and C19) are linked to carbon C4, and an isopropyl group is assigned to carbon C13 [114-116]. It is a very important type of diterpenoid, mainly distributed in the families of Pinaceae, Taxodiaceae, Cupressaceae, Lamiaceae, Araucariaceae, Euphorbiaceae, and so on [117]. They possesse novel and various skeletons. Taiwania quinones owed a 6:5:6 ring system in which the five-membered ring B always carries an aldehyde group being mainly isolated from the Taxodiaceae family [118,119].
A number of this type diterpenoids have been assigned structures that may be derived by rearrangement or cleavage of the abietane skeleton [120], including some of ring A being cleaved [121,122]. Some of them possess a rearranged abietane carbon skeleton of ring A [123,124], and some diterpenes own more highly oxidized abietanes in which ring C is being modified [125]. A few of them formed glycosides [126]. Moreover, some abietane diterpenes are not composed of 20 carbons and only 19 ones and they are called nor diterpenes [127]. The ent‑abietane-type diterpenes are mainly isolated from the genus of Euphorbia [128-130], Isodon [131], and Ceriops [132]. About 700 new abietane-type diterpenes have been reported in the past decade and more details were supplied in Table S5 (Supporting information).
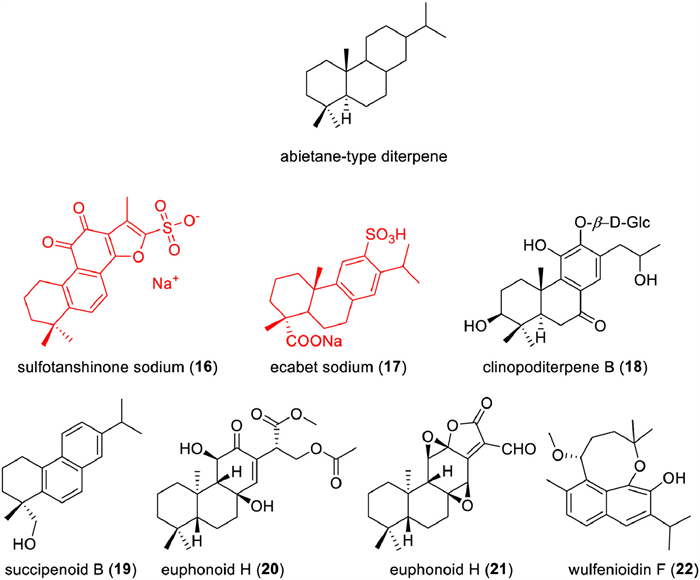
The abietane-type diterpenes own various and complex skeletons displayed a wide spectrum of biological activities, such as treatment of heart disease [133], anti-inflammatory [134-136], cytotoxicities [137-139], β-hematin formation inhibition, neuroprotective [140], α-glucosidase inhibitor [141], antimicrobial activity [142], antidepressant activity [143], against AChE [144]. Some have used in clinical to treat different diseases. Tanshinones are abietane-type diterpene quinones mainly isolated from different Salvia species, especially Salvia miltiorrhiza (Danshen), which is a medicinal plant used in folk medicine for 2000 years that can be considered safe, as well as its components, tanshinones, tanshinone IIA is the most abundant diterpene quinone isolated from S. miltiorrhiza, which has been used in treating cardiovascular diseases for a long time in China [145]. The sodium sulfonate ester (16) of tanshinone IIA has been approved in China since the 1980s as an important treatment for heart disease [146]. Another abietane-type diterpene, ecabet sodium (17), has also been approved in 2010 as an anti-ulcer agent in China (Table 1).
Except these two clinical drugs, some abietane-type diterpenoids displayed various and potent biological activities (Fig. 6), such as clinopoditerpene B (18), a new diterpenoid glycoside, isolated from Clinopodium chinense showed cardioprotective effect against H2O2-induced apoptosis in H9c2 cells [133]; succipenoid B (19), an undescribed nor-abietane diterpenoid, isolated from the CH2Cl2 extract of succinum, showed dose-dependent inhibition of iNOS expression in lipopolysaccharideinduced RAW 264.7 cell [134]; euphonoids H (20) and I (21), two new ent‑abietane diterpenoids, from the ethyl acetate extract of the roots, exhibited significant inhibitory effects against human prostate cancers C4–2B and C4–2B/ENZR cell lines with IC50 values ranging from 4.16 ± 0.42 µmol/L to 5.74 ± 0.45 µmol/L [133]; wulfenioidin F (22), isolated from the whole plant of Orthosiphon wulfenioides, exhibited activity against Zika virus (ZIKV) with median effective concentration (EC50) values of 8.07 µmol/L [147].

|
Download:
|
| Fig. 6. The structural skeleton and the structures of representative abietane-type diterpenoids. | |
{kind=link}
The abietane-type diterpenoids are widely distributed in the world. Due to their moderate to strong and broad range of biological activities, these diterpenoids are interesting experimental objects for further research. Because of their various skeletons and promising biological activities, this type of diterpenoids is a potent source for the leading compounds and clinical drugs.
Acknowledgments: This work was financially supported by the National Natural Science Foundation of China (No. 32100317).
4. ent‑Kaurane-type diterpenoidsCuizhu Wang, Luying Tan, Jinping Liu*
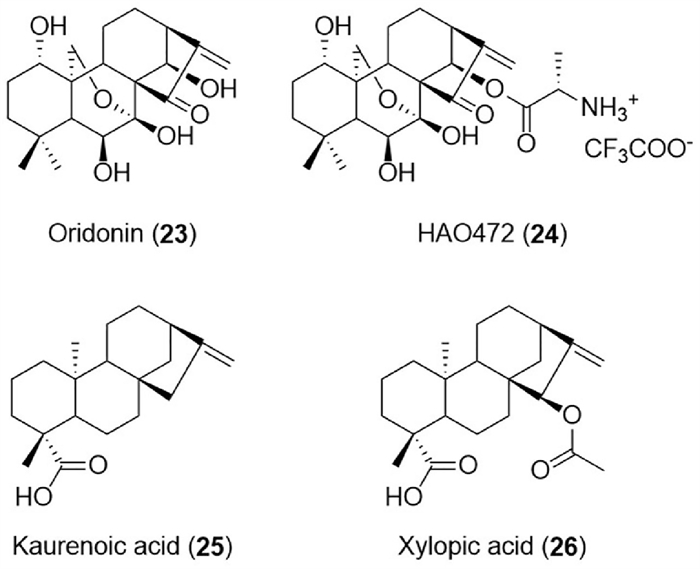
ent‑Kaurane diterpenoids, the enantiomers of kaurane diterpene, were earliest identified from the leaf oil of Kauri pine [148]. The ent‑kauranes are widely distributed in plants (including Asteraceae, Lamiaceae, Euphorbiaceae, Pteridaceae, and Annonaceae families), as well as in liverworts and microbial metabolites. Based on the degree of oxidation and cyclization of the chemical structure, the ent‑kauranes could be divided into C-20 non-oxygenated ent‑kaurane diterpenoids, C-20 oxygenated ent‑kaurane diterpenoids, ent‑6,7-seco-kaurane diterpenoids, and ent‑8,9-seco-kaurane diterpenoids. From 2013 to 2023, a total of 89 ent‑kauranes with various biological activities such as anti-inflammatory [149], antibacterial [150], anticancer [151], antiprotozoal [152], and neuro-protective effects [153] were reported (Table S6 in Supporting information). Among these, oridonin, l-alanine-(14-oridonin) ester trifluoroacetate (HAO472), kaurenoic acid, and xylopic acid have received extensive attention and abundant biological research. Represented ent‑kaurane-type diterpenoids were shown in Fig. 7.

|
Download:
|
| Fig. 7. The structures of representative ent‑kaurane-type diterpenoids. | |
{kind=link}
Oridonin (23), first isolated from Isodon rubescensin in 1967, belongs to C-20 oxygenated ent‑kaurane diterpenoids. It exerted a wide range of biological activities including anti-inflammatory [154], antibacterial [150], antisepsis [155], neuroprotective [156], cardioprotective [157], analgesic [158], immune-modulating [159], antifibrosis [160] and anticancer [161] effects. More than half of the research work was focused on anticancer aspects, mainly prostate cancer [162], lung cancer [163], liver cancer [164], pancreatic cancer [165], ovarian cancer [166], gastric cancer [167], gallbladder cancer [168], oral cancer [169], nasopharyngeal cancer [170], breast carcinoma [171], myeloma [172], osteosarcoma [173], and leukemia [174]. The pathways involved in the anticancer effect have also been elucidated. For example, the phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT) and p53 pathways in anti-prostate cancer [175], PI3K/AKT/mammalian target of rapamycin (mTOR) and Ras/Raf pathways in anti-esophageal cancer [176], reactive oxygen species (ROS)/c-Jun N-terminal kinase (JNK)/c-Jun axis in anti-colorectal cancer [177], epithelial-mesenchymal transition (EMT) and the hypoxia inducible factor-1 alpha (HIF-1α)/vascular endothelial growth factor (VEGF) signaling pathway in anti-breast cancer [178]. In the evaluation of druggability, the pharmacokinetic parameters after gavage administration of oridonin (40 mg/kg) in rats were also obtained by using liquid chromatography-tandem mass spectrometry (LC-MS/MS) technology [179]. The main parameters were as follows: t1/2 was 10.88 ± 4.38 h; Tmax was 1.00 ± 0.12 h; Cmax was 146.9 ± 10.17 ng/mL; AUC0–t was 1.31 ± 0.29 mg h L−1. Additionally, oridonin was mainly metabolized in the liver tissue. According to the report, oridonin has a solubility of 0.75 mg/mL and a logP of 1.66, and led to a disappointing oral absolute bioavailability of 0.8%–4.32% [180]. As for the toxicity and safety evaluation of oridonin, there was evidence that it might exhibit hepatotoxicity under specific circumstances [181]. Namely, the hepatic cord was narrowed and the alanine aminotransferase (ALP) level was significantly increased in nude mice treated with 10 or 5 mg/kg of oridonin. To sum up, although oridonin had good biological activity, poor safety and low bioavailability have limited its further clinical application. The study on the SAR of oridonin showed that the conjugated pentatomic ring may be the anti-tumor active group, including the α-methylene-cyclopentanone system. To improve the drug-like properties of oridonin, a series of oridonin derivatives were prepared.
As a result of the structural optimization of oridonin (the lead compound), l-alanine-(14-oridonin) ester trifluoroacetate (HAO472, 24) was acquired. HAO472 was then progressed to Phase Ⅰ clinical trial by Hengrui Medicine Co. Ltd., in China for the treatment of acute myelogenous leukemia (80–320 mg/d, iv) [162]. In fact, HAO472 could be considered a prodrug due to its metabolizing to the parent compound oridonin in vivo by the cleavage of C-14 ester bond. The research results indicated that HAO472 had similar pharmacological activities to oridonin, but its toxicity and vascular injury were significantly reduced [182]. In addition, some preclinical studies on the pharmacological activity of HAO472 have also been carried out. The results showed that it could ameliorate 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis by modulating the subsets and functions of lymphocytes, suppressing inflammation, and inhibiting the nuclear translocation of NF-κB p65 subunits in mice with colitis [182]. HAO472 was also reported to possess remarkable binding selectivity to coronavirus disease 2019 (COⅥD-19) and HCoV-229E Nsp9 over FSCoV-F56 Nsp9, suggesting that HAO472 could be active against coronavirus [183]. Moreover, a series of HAO472 amino acid ester derivatives were synthesized and were screened to have high efficiency and selectivity against SGC-7901, Bel-7402, HL-60, PC-3, A549, and K562 cancer cell lines, inspiring further research [184].
Kaurenoic acid or kaurenic acid (KA, 25) commonly existed in medicinal plants such as Acanthopanax trifoliatus and Acanthopanax gracilistylus. KA possesses multiple activities such as anti-inflammatory [185], analgesic [186], antibacterial [187], antitumor [188], hepatoprotective [189], antiparasitic [190], anticonvulsant [191], antisteatosis [192], antituberculosis [193], hypoglycemic [194] and antiasthma [195] effects. Furthermore, Nrf2, TGF-β signaling, Th2, and NF-κB/cytokine-related pathways have been confirmed to be involved in the anti-inflammatory activity of KA [185]. The pharmacokinetic profile of KA has also been determined [196]. The results showed that linear and two-compartment kinetic behavior were indicated after intravenous administration of KA (50 mg/kg). The main parameters were as follows: Cmax=22.17 ± 1.65 mg/L, CL=17.67 ± 1.50 mL min kg−1, AUC0-∞=2859.65 ± 278.42 mg min L−1, t1/2 = 9.52 ± 0.61 h, and Vd=14.53 ± 1.47 L/kg, suggesting that KA with liposolubility was widely distributed in body tissues and fluids. However, the plasma level of KA (50 mg/kg) by oral administration could not be detected by using high performance liquid chromatography-ultraviolet detector (HPLC-UV) method, indicating poor absorption or extensive pre-systemic elimination.
Xylopic acid (XA, 15β-acetoxy‑ent-kaur-16-en-19-oic acid, 26) was isolated from the fruit of Xylopia aethiopica (Annonaceae) [197]. It has wide pharmacological activities such as anti-anxiety [198], antidepressant-like [199], analgesic [200], anti-inflammatory [197], anti-allergic [201], anti-cancer [202], anti-malarial [203] effects. In particular, the anti-inflammatory and neuroprotective effects received more attention. For instance, it has been found that XA played its pharmacological role in acute inflammation by effectively modulating pro-inflammatory markers including prostaglandin E2, serotonin, histamine, and bradykinin [197]. It was further elucidated that the mechanism of XA’s anti-inflammatory effect might be due to its ability to modulate the activity of Nrf2 and NF-κB [204]. In terms of chemical composition research, the detailed imaging and quantitative characterization of XA were performed by using MALDI-HRMS imaging and HPLC—HRESI-MSn technology, respectively [205]. As for the pharmacokinetic study in vitro (in rats) and in vivo (in rat liver microsomal enzyme), the main parameters of oral administration XA (100 mg/kg) were as follows: Cmax=167.03 ± 6.18 ng/mL, t1/2 = 13.03 ± 7.33 h, CL=0.04 ± 0.01 mL h kg−1, mean residence time (MRT) =23.83 ± 11.02 h. Furthermore, a total of six metabolites were tentatively identified after subjecting XA to rat liver microsomal enzyme metabolism [206].
In summary, the natural ent‑kaurane diterpenoids had diverse structures and could provide abundant resources for the research and discovery of new drugs. The unique chemical structural skeleton and the stereochemistry generated specific activities and exhibited important roles in life processes by interacting with various drug targets. Consequently, the ent‑kaurane diterpenoids could be considered a group of lead compounds with unique chemical structures and various pharmacological activities. In the future, optimization of structure is suggested to be carried out aiming at obtaining derivatives with more satisfactory lipid water distribution coefficient, higher oral bioavailability, stronger biological activity, and less toxic side effects.
Acknowledgment: This research was funded by the Key Research and Development Program of Jilin Province (Nos. 20230204038YY, 20240305052YY).
5. Grayanane-type diterpenoidsLixin Zhao, Hongye Han, Yong Li*
Grayanane-type diterpenoids, exclusively found in plants of the Ericaceae family, are well-known toxic constituents in genera such as Rhododendron, Pieris, Leucothoe, Craibiodendron, Lyonia, and Kalmia. However, they also hold great potential for disease treatment. To date, over 400 grayanane and related derivatives have been identified from natural sources, exhibiting a diverse range of structural variations and many displaying significant biological activities (Table S7 in Supporting information).
A multitude of grayanane diterpenoid skeletons have been consistently uncovered from ericaceous species over the past decade (as highlighted in Fig. 8). These include 5,6-secograyanane [207], d-homograyanane (featuring a 5/7/6/6-fused ring system) [208], mollane (C-nor-d-homograyanane) [209], 1,10:2,3-disecograyanane. [210], 2,3:5,6-disecograyanane [211], rhomollane (featuring a 5/6/6/5 tetracyclic system) [212], rhodomollane (featuring a 5/7/5/5 tetracyclic system) [213], and mollebenzylane [214], among others.

|
Download:
|
| Fig. 8. Novel grayanane-related skeletons and representative compounds uncovered from ericaceous species over the past decade. | |
{kind=link}
These novel and complex structures not only provide inspiration and challenges for synthetic chemists but, more importantly, serve as a valuable source for the discovery of potential lead compounds in drug development. Herein, we summarize the recent advancements in the discovery of highly active compounds within this class of diterpenes over the past decade and discuss their pharmacological properties, highlighting their potential as drug candidates.
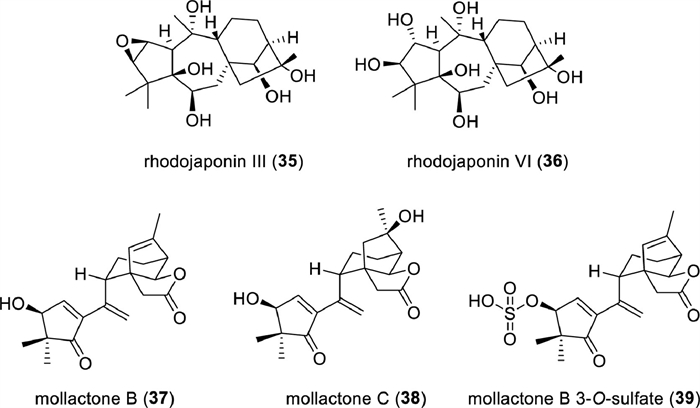
Given the traditional applications of various plants in the Ericaceae family for analgesic and anesthetic purposes, it is logical to explore and discover lead compounds with analgesic properties from this source. Among them, a variety of grayanane diterpenoids have shown significant antinociceptive effects in the acetic acid-induced writhing test, as evidenced by Table S7 provided in supporting information. Particularly noteworthy were rhodojaponins Ⅲ (35) and Ⅵ (36) (Fig. 9), which exhibited remarkable potency with median inhibitory dose (ID50) values as low as 46.9 and 72.7 µg/kg, respectively. These two compounds demonstrated superior potency over morphine across multiple pain models, including acute, inflammatory, and neuropathic pain [209].

|
Download:
|
| Fig. 9. The structures of representative high-activity grayanane diterpenoids. | |
{kind=link}
The current treatment options for neuropathic pain are often inadequate, highlighting the urgent need for new lead compounds especially those with new therapeutic targets. Rhodojaponin Ⅵ (36), as a lead compound for neuropathic pain, was investigated for its direct target and mechanism of action (MOA) by Yu and coworkers. N-Ethylmaleimide-sensitive fusion (NSF) has been identified as the primary direct target of rhodojaponin Ⅵ through thermal proteome profiling and subsequent validation experiments. It was found that NSF facilitated the trafficking of Cav2.2 channels, increasing in Ca2+ current intensity associated with neuropathic pain. Remarkably, rhodojaponin Ⅵ was able to reverse the effects of NSF, suggesting its potential as a unique class of analgesic natural product targeting Cav2.2 channels via NSF. This study sheds light on a novel mechanism for treating neuropathic pain and provides valuable insights for future research and development of therapeutic interventions [215].
Mollactones A–C (33, 37, 38), three 5,6-seco-grayanane diterpenoids with a unique 3-oxa-tricyclo[4,3,2,02,6]undecane motif, were isolated from R. mole by Yao and colleagues [207]. These compounds displayed significant protein tyrosine phosphatase-1B (PTP1B) inhibitory activity in a competitive mode, with IC50 values of 4.24 ± 0.21, 2.69 ± 0.23, and 3.33 ± 0.22 µmol/L, respectively. Mollactone B 3-O-sulfate (39) was designed based on molecular docking studies and exhibited improved inhibitory activity against PTP1B (IC50=0.22 ± 0.05 µmol/L, Ki=6.79 ± 1.28 µmol/L) (Fig. 9). These findings provide insights for designing novel PTP1B inhibitors aimed at combating both obesity and type Ⅱ diabetes mellitus.
Acknowledgment: This work was supported by grant from the National Natural Science Foundation of China (No. 21977119).
6. Ingenane-type and tigliane-type diterpenoidsLiefeng Ma, Zhajun Zhan*
Ingenane diterpenoids possess a 5/7/7/3-tetracyclic ring system featuring an extremely rare trans-intrabridgehead bicyclo[4.4.1]undecane, usually bearing a ketone group at C-9. To date, ingenane diterpenoids are only found in plants of Euphorbiaceae family, especially the genus of Euphorbia [216]. Ingenane diterpenoids usually occur in esterified forms as the irritant, vesicant, and tumor-promoting constituents of the Euphorbia plants. The acyl groups are generally aliphatic, sometimes extensively unsaturated. The acyloxy groups are usually found at C-3, C-5, and C-20, but in some cases, they can be found at C-13 and C-17 [5,217].
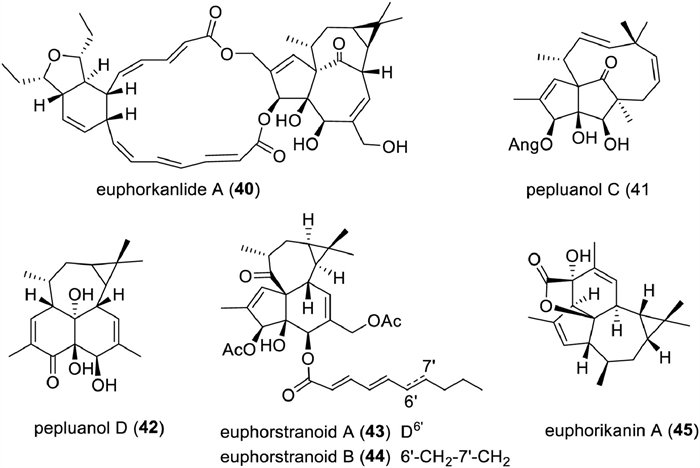
In the past decade, 55 new ingenane diterpenoids have been reported from plants, including several novel ones with unprecedented skeletons. Their detailed information, including trivial names, structures, sources, and bioactivities, is shown in Table S8 (Supporting information). Euphorkanlide A (40) is a highly modified ingenane with a C24 appendage forming an additional 5/6/19 ring system. The key biosynthetic step for generating this ring system was postulated to proceed through an intramolecular Diel-Alder reaction between two polyene side chains [218]. Compounds 41–45 are six rearranged ingenane diterpenoids (Fig. 10). Pepluanol C (41) features a 5/5/10 ring system incorporating a [7.2.1]bicylcododecane moiety, while pepluanol D (42) possesses a new 6/6/7/3 fused-ring skeleton. Biosynthetically, they might originate from 20-deoxyingenol 3-angelate, an abundant component in Euphorbia peplus, with a 1,3-shift rearrangement and a retro-aldol reaction as key biosynthetic step, respectively [219]. Euphorstranoids A and B (43, 44) might be rearrangement products of a co-occurring ingenane diterpenoid euphstrachenol C via two consecutive 1,2-carbon shift, and the hypothesis was supported by chemical transformation [220]. Euphorikanin A (45) from the roots of E. kansui is a novel 5/6/7/3-fused tetracyclic lactone, which might be derived from 4-O-acetyl-5-O-benzoyl-3β‑hydroxy-20-deoxyingenol with enzymatically catalyzed retro-aldol and aldol reactions as key steps [221].

|
Download:
|
| Fig. 10. The structures of representative rearranged ingenane diterpenoids. | |
{kind=link}
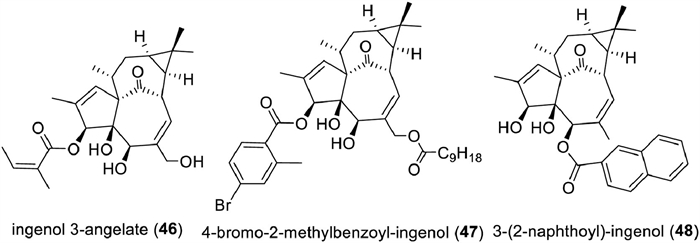
Ingenane diterpenoids have a wide range of therapeutically relevant bioactivities, such as antitumor [222-224], multidrug resistance reversal [225,226], antiviral [227-231], and anti-inflammatory activities [232,233], of which antitumor and anti-HIV drew the most attention. Ingenol 3-angelate (ingenol mebutate, PEP005, Picato, LEO Pharma, 46) has been approved by the FDA in 2012 to treat actinic keratoses [234,235]. Recent mechanistic study on ingenol mebutate revealed that its target was the mitochondrial carnitine acylcarnitine translocase SLC25A20 [236]. Ingenane diterpenoids not only selectively inhibit HIV replication, but also reactivate latent HIV-1 due to their PKC activation potential [231,237]. To avoid the bolus toxicities of ingenol esters, a series of analogs were synthesized. Several analogs with better drug-ability were afforded, such as 4‑bromo-2-methylbenzoyl-ingenol (47), and 3-(2-naphthoyl)-ingenol (48) [238,239]. These studies also revealed the acyl group at C-3 was a key element for the retention or improvement of the anti-HIV activity (Fig. 11).

|
Download:
|
| Fig. 11. The structures of representative ingenane-derived drugs or drug candidates. | |
{kind=link}
In the time span of 2013−2023, only two ingenane diterpenoids were totally synthesized, namely ingenol [240,241] and euphorikanin A [242]. In 2013, an efficient and highly stereo-controlled synthesis of (+)-ingenol was completed in only 14 steps from inexpensive (+)−3-carene [238]. This synthesis highlights a two-phase approach and a key pinacol rearrangement reaction. The total synthesis of euphorikanin A was reported in 2021 also with (+)−3-carene as the tarting material, and this synthesis highlights a SmI2-mediated ketyl-enoate reaction to construct the 3/6/5 ring system in euphorikanin A [243]. For more detailed information, the readers can read comprehensive reviews recently published [5,216,244].
Tigliane diterpenoids are based on a 5/7/6/3-tetracyclic ring system with the feature of an α,β-unsaturated ketol in the A ring. Tigliane diterpenoids usually possess polydroxy groups at their C-12, C-13, and C-20 positions which can be readily esterified by acetic, isobutyric, tiglic, 2-methylbutyric, benzoic, 2-methylaminobenzoic, or saturated and unsaturated long-chain aliphatic fatty acids [244,245]. The distribution of tigliane diterpenoids was limited to plant species belonging to the Euphorbiaceae and Thymelaeaceae families. The genera Euphorbia, Croton, Jatropha and Trigonostemon of Euphorbiaceae family, and the genera Daphne, Stellera, Wikstroemia and Homalanthus of Thymelaeaceae family are rich sources of this type diterpenoids [85,217,244-246].
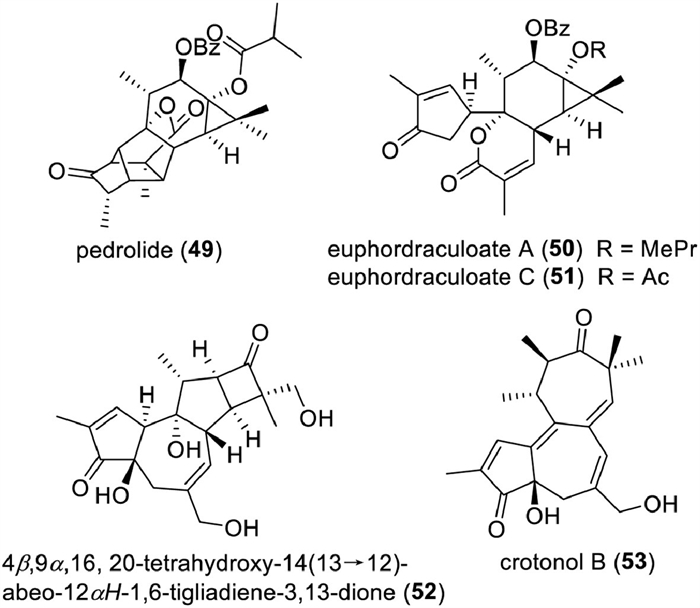
As shown in Table S9 (Supporting information), there are 154 new tigliane diterpenoids are reported in this period. Tigliane diterpenoids would likely exist in the form of aglycons, and only a few tigliane glycosides have been reported in this period [247-251]. Compounds 49∼53 are four rearranged tigliane diterpenoids with unprecedented scaffolds (Fig. 12). Pedrolide (49), a cage-like diterpenoid containing an unusual bicyclo[2.2.1]heptane system, was isolated from Euphorbia pedroi as a P-glycoprotein inhibitor. Biosynthetically, it was proposed to be derived from a tigliane with an intramolecular Michael addition and 1,2-alkyl shift rearrangement as key steps [252]. Euphordraculoate A (50) [253] and euphordraculoate C (51) [254] are two 4,5-seco-tigliane lactones, which were isolated from E. dracunculoides and E. usambarica, respectively. A retro-aldol and an intramolecular esterification were proposed to be involved in their biosynthesis. Chemical research on the roots of E. ebracteolata afforded a rare 14(13→12)-abeo-tigliane diterpenoid (52) [251]. Crotonol B (53), a 13,14-seco-tigliane with a 5/7/7-fused ring core, was characterized from Croton tiglium as a cytotoxic constituent [255].

|
Download:
|
| Fig. 12. The structures of representative rearranged tigliane diterpenoids. | |
{kind=link}
Tigliane diterpenoids are famous for their PKC-modulating effects and usually possess “negative” bioactivities, such as tumor-promoting, inflammatory, and skin-irritant activities [245]. Phorbol 12-myristate 13-acetate (TPA, 54), a potent PKC activator, has been used as a pharmacological and biochemical tool to induce inflammation and investigate tumor promotion mechanisms (Fig. 13). Interestingly, some tigliane diterpenoids exhibit “positive” biological activities such as anti-cancer, anti-HIV, nematicide, and neurogenesis with no tumor- or inflammation-promoting activities [229,256-261]. This difference in activity is thought to be due to the selectivity of these diterpenoids for the protein kinase C (PKC) isozymes [262]. Most recently, tigilanol tiglate (Stelfonta, 55) was approved by FDA for the treatment of dogs with non-metastatic, cutaneous mast cell tumors. Due to its efficacy, it is also being evaluated for human cancer treatment [263]. Furthermore, compound 55 and its analogs could stimulate skin keratinocyte wound healing responses and re-epithelialization, and induce biofilm disruption via the activation of PKC. Thereby, they can be used as a novel class of topical therapeutics for chronic wounds, such as non-healing skin wounds, and are now in clinical development [264,265]. Tigliane diterpenoids display dual activity against HIV-1, not only inhibiting its replication but also activating the latent HIV-1 in CD4+ T cells. This indicates their potential as latency-reversing agents (LRAs) for curing HIV infection based on a “shock and kill” strategy [238,266]. SAR studies revealed that their potency and selectivity are closely associated with the type of acyls and their substituted position [267,268]. Phorbol 12,13-diisobutyrate (56) could facilitate the release of TGFα possibly by activating PKCδC1B, leading to promote neural progenitor cell proliferation, and has the potential to treat disorders associated with a reduction in neurogenesis and memory impairment [269].

|
Download:
|
| Fig. 13. The structures of representative tigliane-derived drugs or drug candidates. | |
{kind=link}
Recently, tigliane diterpenoids were revealed to be lipid-lowering agents for anti-obesity drug development. 12-O-Benzoyl-13-O-[2-methylpropanoyl]−4,20-dideoxy-5-O-acetylphorbol (57) significantly retarded the differentiation of 3T3-L1 adipocyte by inhibiting the glucocorticoid receptor α-Dexras1 axis [270].
In the time span of 2013−2020, several new synthetic strategies have been developed to construct 5/7/6 tricyclic system in tiglianes, including transannular intramolecular aldol reaction [271], oxidative dearomatization [272], Pauson-Khand reaction [273], and furan-allene [4 + 3] cycloaddition [274,275]. Based on these strategies, the total synthesis of two tiglianes, namely phorbol and prostratin, has been achieved [177]. For detailed information on these syntheses, the readers can refer to a comprehensive review published in 2021 [243]. In this review, we only supplement the new entries to this class by introducing the latest work. In 2021, Inoue’s group developed a new, unified strategy for expeditious total syntheses of polycyclic diterpenoids bearing a 5/7/6-tricyclic ring system. This strategy includes two stages, the synthesis of an advanced common intermediate (ACI) and the derivatization of ACI to the target molecules, in which the assembling of the 5/7/6 core of ACI highlighted the reliability of the stereo-chemically predestined and highly reactive bridgehead radical for construction of a densely functionalized compound [276]. In 2023, Carreira reported the first total synthesis of (+)-pedrolide (49). The total synthesis highlights the construction of the bicyclo[2.2.1]heptane core via an intramolecular cyclopentadiene-Diels−Alder cycloaddition with a norbornadiene serving as an effective surrogate for cyclopentadiene [277].
7. Daphnane-type diterpenoidsKouharu Otsuki, Wei Li*
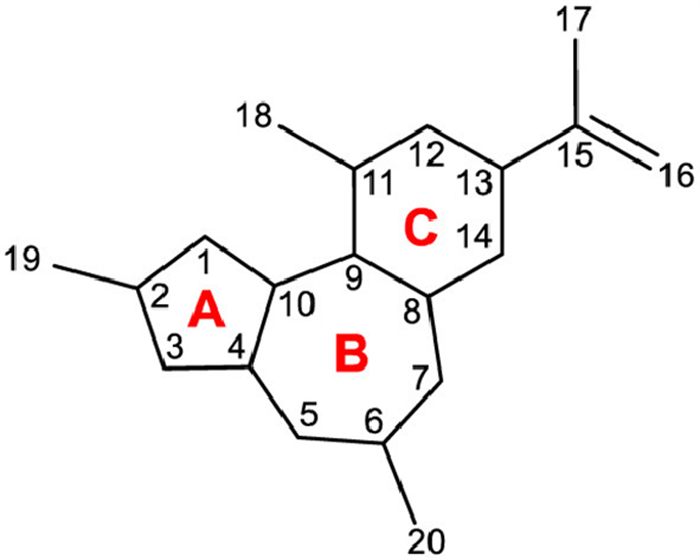
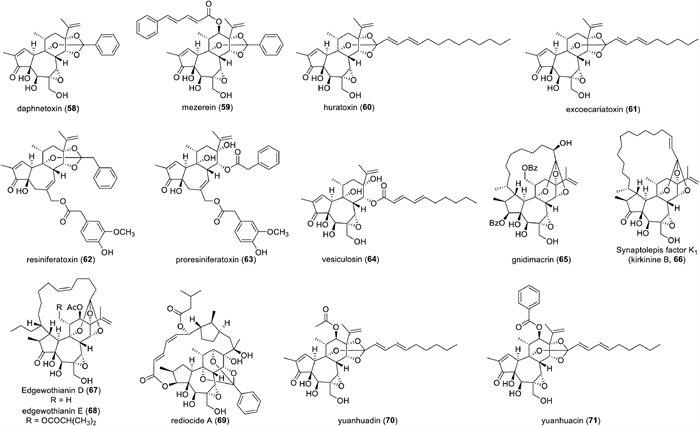
Daphnane diterpenoids have a trans-fused 5/7/6 (A/B/C) tricyclic structure, characterized by an isopropenyl group attached to C-13 at the six-membered ring (C-ring) (Fig. 14). Over the past half century, this type of diterpenoids is found in plants of the Thymelaeaceae and Euphorbiaceae families. In 1970, the first daphnane diterpenoid, daphnetoxin (58), was isolated as a potent toxic compound from Daphne mezereum (Thymelaeaceae) (Fig. 15) [278]. Around the same time, mezerein (59) was also isolated from D. mezereum, and later, in vitro and in vivo experiments demonstrated that it exhibited potent anti-leukemic activity, leading to increased attention on daphnane diterpenoids as potential anticancer agents [279,280]. On the other hand, in 1971, huratoxin (60) was isolated with potent piscicidal activity from Hura crepitans (Euphorbiaceae), revealing the presence of daphnane diterpenoids in plants of the Euphorbiaceae family [281,282]. Daphnane diterpenoids represented by compounds such as 58–60 are characterized by an orthoester acylate formed at C-9, C-13, and C-14 of the C-ring and possess an α,β-unsaturated ketone group in the A-ring and a 4β,5β-dihydroxy-6α,7α-epoxy structure in the B-ring. Resiniferatoxin (61), a promising drug candidate among the daphnane diterpenoids for pain treatment, possesses a 9,13,14-orthoester structure but lacks a 5β‑hydroxy group and a 6α,7α-epoxy group, instead having the 6,7-ene structure commonly found in the phorbol esters. Compounds with the 6,7-ene structure in the B-ring have been reported only from plants of the genus Euphorbia since the isolation of 4 from Euphorbia resinifera and E. unispina in 1975 [283]. Proresiniferatoxin (62), isolated alongside 63 from E. resinifera and E. unispina, as well as vesiculosin (64), isolated with excoecariatoxin (65) from Diarthron vesiculosum (Thymelaeaceae) [284], have a 9,13,14-trihydroxy structure resulting from the cleavage of the 9,13,14-orthoester moiety. Since 62 readily converts to 61 under high temperature or acidic conditions, compounds with a 9,13,14-trihydroxy structure are proposed as precursors in the biosynthesis of compounds with a 9,13,14-orthoester structure [285].

|
Download:
|
| Fig. 14. Skeletal structure of daphnane diterpenoid. | |
{kind=link}

|
Download:
|
| Fig. 15. Structure of representative daphnane diterpenoids isolated from plants of the Thymelaeaceae and Euphorbiaceae families. | |
{kind=link}
Additionally, daphnane diterpenoids occur both in compounds with and without a substituent attached to the C-12 of the C ring, such as 58 and 59. Compounds with the above-mentioned multiple oxygenated functionalities in the diterpene skeleton are the most universal of the naturally occurring daphnane diterpenoids.
Furthermore, gnidimacrin (65), isolated with potent anti-leukemic activity from the Gnidia subcordata (Thymelaeaceae) in 1976, possesses a chemical structure characterized by an aliphatic chain with an orthoester linkage to C-9, C-13, and C-14 is connected to C-1 of the A-ring, forming a macrocyclic ring that spans the diterpene skeleton [286]. Since the discovery of 64 from the genus Gnidia, compounds with similar macrocyclic structures have been reported to be isolated from various genera within the Thymelaeaceae family, including Daphne, Dphnopsis, Dirca, Edgeworthia, Pimelea, Stellera, Synaptolepis, and Wikstroemia [245]. The macrocyclic daphnane diterpenoids isolated from the Thymelaeaceae family are generally possessed a macrocyclic ring originated from a C10 aliphatic chain. As a variant of these daphnane diterpenoids, synaptolepsis factor K1 (also known as kirkinine B, 66), featuring a macrocyclic ring formed from a C16 aliphatic chain, was isolated from plants of the genus Synaptolepis in 1977 [287-289]. Furthermore, compounds with a macrocyclic ring formed from a C14 aliphatic chain, such as edgeworthianins E (67) and D (68), were reported to be present in Edgeworthia chrysantha in 1983 [290,291]. In 2000, a highly modified daphnane diterpenoid, rediocide A (69), with potent insecticidal activity, was isolated from the Trigonostemon reidioides (Euphorbiaceae) [292]. 69 is characterized by the formation orthoester acylate at C-9, C-12, and C-14 of the C-ring, and a highly modified unsaturated acyl chain attached to C-3 is connected to C-16 of the isopropyl group, forming a macrocyclic ring that spans the diterpene skeleton. This type of macrocyclic daphnane diterpenoids has been isolated exclusively from the genus Trigonostemon.
Since daphnane diterpenoids initial discovery in the 1970s, they have been continuously investigated, resulting in the isolation of over 350 compounds from various plants to date. Liao et al. summarized daphnane diterpenoids isolated from the Thymelaeaceae and Euphorbiaceae families up to 2009 [293]. The daphnane diterpenoids isolated after that were listed in Table S10 (Supporting information).
Daphnane diterpenoids exhibit structurally diverse due to the presence of multiple oxygenated functionalities in the polycyclic diterpene skeleton and have also been found to have a variety of attractive biological activities, including analgesic, anticancer, and anti-HIV activities. Resiniferatoxin (61) has garnered significant attention in drug discovery since its capsaicin-like activity was discovered. 61 shares the vanillyl substituent, a structural motif essential for the expression of the bioactivity of capsaicin. Consequently, it acts as an agonist of the transient receptor potential vanilloid 1 (TRPV1; also known as capsaicin receptor), serving as a capsaicin analog with three to four orders of magnitude more potent activity than capsaicin [294]. 61 has played a crucial role not only in understanding the function of TRPV1 but also as a lead compound for developing TRPV1 agonists or antagonists. Additionally, it shows promise as a potential analgesic agent, exhibiting analgesic effects by inducing desensitization of capsaicin-sensitive nerves through potent TRPV1 stimulation [295,296]. Phase Ⅱ and Ⅲ clinical trials of 61 have been conducted for the management of cancer pain and knee osteoarthritis-related pain, aiming to develop an analgetic drug without the side effects and physical dependence associated with opioid analgesics [297]. Recently, 61 received Breakthrough Therapy Designation from FDA for pain associated with osteoarthritis of the knee and is expected to accelerate clinical development and marketing approval of 61 in the future.
On the other hand, daphnane diterpenoids have attracted attention for their anticancer and anti-HIV activities, and their antiproliferative activity against various cancer cell lines and inhibition of HIV-1 replication have been investigated. Research on the anti-cancer activity of daphnane diterpenoids has been active since the mid-1970s, and so far naturally occurring daphnane diterpenoids have been reported to exhibit anti-cancer activity against various types of cancer cells, including leukemia, non-small cell lung cancer, melanoma, hepatocellular carcinoma, triple-negative breast cancer, and castration-resistant prostate cancer [298-304]. Recently, research has been progressing on the molecular mechanisms of anti-cancer activity of compounds such as yuanhuadin (70) and yuanhuacine (71) [305,306]. Yuanhuadin (70) has been reported to regulate checkpoint proteins, inducing the up-regulation of p21 and the down-regulation of cyclins, cyclin-dependent kinases 2 (CDK2) and 4 (CDK4), as well as c-Myc. 70 also suppressed the expression of the Akt/ mTOR, EGFR, and c-Met-signaling pathways. Yuanhuacine (71) has been reported to trigger various cellular effects, inducing up-regulation of checkpoint proteins such as p21 and p38. Additionally, 71 was found to regulate the AMPK/mTOR signaling pathway by suppressing the activation of mTORC2-associated downstream targets including Akt, PKCa, Rac1, and F-actin.
Research on anti-HIV activity has been actively conducted since the 2010s. Results evaluating the inhibition of HIV-1 replication of naturally occurring daphnane diterpenoids, including daphnetoxin (58) and excoecariatoxin (61), demonstrated more potent activity than approved HIV drugs (reverse transcriptase inhibitors and protease inhibitors) [307]. The suggested mechanism involves the direct inhibition of HIV-1 co-receptors CCR5 and CXCR4. Gnidimacrin (65) exhibited approximately 1000 times more potent HIV replication inhibition and latent HIV-1 activation than prostratin, a tigliane diterpenoid that underwent clinical trials as an anti-HIV candidate [308]. Studies on the mechanism of action of 65 suggest that the activation of latent HIV-1 transcription through selective activation of PKCβ1 and PKCβ2 is involved in the expression of anti-HIV activity [309].
Daphnane diterpenoids have been reported to exhibit excellent anti-cancer and anti-HIV activities, however, no daphnane diterpenoid has reached clinical application yet. Further validation of the activity of daphnane diterpenoids in vivo models and pharmacokinetic studies is desirable for the development of novel therapeutic agents against cancer and HIV infections.
Acknowledgment: This work was supported by the Japan Society for the Promotion of Science (No. KAKENHI 21K06619).
8. Aconitine-type diterpene alkaloidsXianli Zhou*
Diterpenoid alkaloids (DAs), originating from the amination of natural tetracyclic diterpenes, are a special category of natural products with complex cyclic skeletons and remarkable pharmacological profile mainly involving analgesic, anti-inflammatory, anti-arrhythmic, and cardiotonic properties, however infamous toxicity as well. Their history can be traced back to 1833, when the first DA, aconitine, was isolated from Aconitum napellus L. by P.L. Geige [310]. To date, over 1500 DAs have been isolated and characterized [311]. Four representative DAs, 3-acetylaconitine [312], bulleyaconitine A [313], lappaconitine [314], and acehytisine (Table S11 in Supporting information) [315], have now been clinically approved. Furthermore, mesaconine [316] has been researched under clinical studies for its marked cardiac activities. Therefore, DAs have attracted continuing attention from both chemists and biologists.
This part of the review extensively summarizes a total of 265 naturally occurring DAs reported from the plant kingdom from 2013 to 2023, taking into account their distributions, structural classifications, and bioactivities. It provides a comprehensive and in-depth perspective for further investigation on DAs.
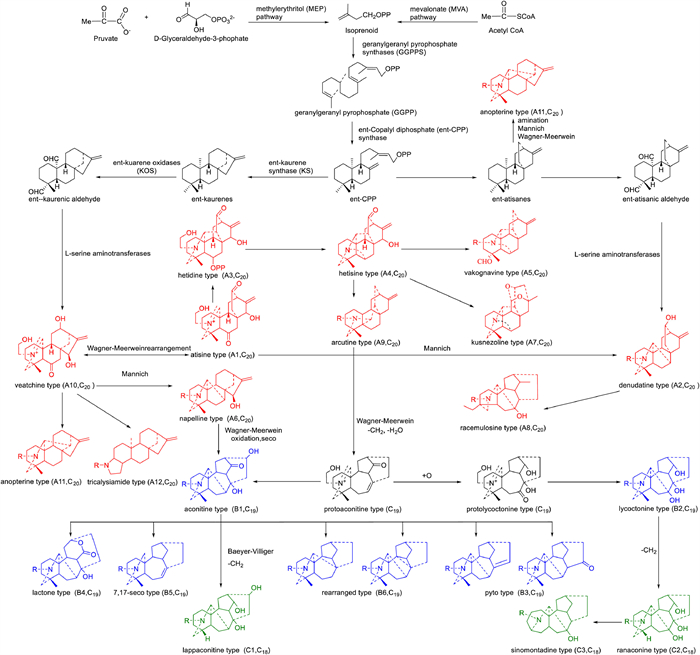
The occurrence of DAs throughout the Ranunculaceae species is idiosyncratic. Ranunculaceae species, especially the genera Aconitum, Delphinium, and Consolida, are found to be the main sources of C19- and C20-DAs while the genera Aconitum are the primary source of C18-DAs. Besides, partly C20-DAs are discovered in the genera Spiraea of Rosaceae species. In this review, a modified DAs category system covering C18-, C19-, C20- and bis-DAs since 2009 [317] is used to classify Das (Fig. 16).

|
Download:
|
| Fig. 16. The structural skeletons of diterpenoid alkaloids. | |
{kind=link}
C20-DAs are likely the precursor of the C18- and C19-DAs. C20-DAs are complex and diverse, with the 12 common structural classes of atisine type (A1), denudatine type (A2), hetidine type (A3), hetisine type (A4), vakognavine type (A5), napelline type (A6), kusnezoline type (A7), racemulosine type (A8), arcutine type (A9), veatchine type (A10), anopterine type (A11), and tricalysiamide type (A12). The carbon skeleton of atisine type DAs is identical to atisine diterpenes, and it is considered to be the original group. Denudatine type DAs are a class of hexacyclic C20-DAs based upon the atisine class with an extra bond between C-7 and C-20. Hetidine class DAs are similar to the atisine class DAs except for a hexacyclic bearing the additional C-7–C-20 bond. Hetisine type DAs possess the additional C-14–C-20 bond and N–C-6 bond in the atisine class. Vakognavine class DAs are characteristic of the hexacyclic system with N, C20-seco hetisine type. Napelline class DAs are similar to veatchine class except for an additional C-20–C-7 bond. Kusnezoline type DAs possess two tetrahydropyran rings in structure. The A ring of racemulosine type is rearranged. Arcutine type DAs are nearly identical to atisine type DAs except for an additional C-5–C-20 bond. The carbon skeleton of veatchine class DAs is the same as the kaurane diterpenes. The carbon skeleton of anopterine type is identical to the veatchine class diterpenes except for an extra C-20–C-14 bond. Tricalysiamide type DAs are the rearranged skeleton of the veatchine class DAs with an additional C-3–C-19 bond.
C19-DAs are the most abundant class of DAs and nearly 1000 natural C19-DAs have been found so far. They are generally divided into 6 classes: aconitine type (B1), lycoctonine type (B2), pyro type (B3), lactone type (B4), 7,17-seco type (B5), and rearranged type (B6). Over 50% of the reported C19-DAs are aconitine-type DAs. Lycoctonine class DAs are characterized by an extra oxygenated group at C-7 compared to aconitine type DAs. The skeleton of pyro-type DAs possesses a double bond between C-8 and C-15 or a carbonyl at C-15. Lactone class DAs feature a six-membered lactonized C ring. The C-7–C-17 bond is broken in 7,17-seco type DAs. Acoseptine subtype of rearranged type DAs features an additional C-8–C-17 bond in the structure. Vilmoraconitine subtype of rearranged type possesses a three-membered ring of C-8–C-9–C-10.
C18-DAs are mainly classified into 3 classes: lappaconitine (C1), ranaconine (C2), and sinomontadine types (C3). When the C-18 of aconitine type DAs is oxidatively degradated, they become lappaconitine type DAs. Ranaconine-type DAs feature an additional hydroxy at C-7 compared to lappaconitine-type DAs. Sinomontadine-type DAs possess seven-membered A ring.
Bis-DAs are a class of relatively small but special components in DAs. They are usually divided into 6 classes: atisine–denudatine type, hetidine–hetisine type, denudatine–denudatine type, atisine–hetidine type, rearranged atisine–hetidine type, heteratisine–denudatine type, heteratisine–hetidine type.
Since 2013, 265 DAs, including 54 C20-DAs, 189 C19-DAs, 15 C18-DAs, and 7 bis-DAs, have been isolated and identified from plants mostly in the Ranunculaceae family. Numerous alkaloids have significant preclinical effects including analgesic, anti-arrhythmic, anti-inflammatory, muscle relaxant, hypotensive, anti-bacterial, anti-tumor, anti-feedant activities, and so on. Part of these compounds exhibited significant activities and showed potential to be leading compounds.
As shown in Fig. 17, aconicatisulfonines A (72) and B (73) [318] exhibited remarkable analgesic activities against acetic acid-induced mice writhing in a dose-dependent manner, while 73 showed the highest analgesic inhibition of 78.34%. Taipeinine A (74) [319] potently suppressed the proliferation of cells HL-60 and K562 with IC50 values of 0.2 ± 0.05 mg/mL and 0.7 ± 0.15 mg/mL, respectively. In the anti-inflammatory activities, the ability of szechenyianine B (75) [320] to inhibit the NO production on LPS-activated RAW264.7 cells was stronger than the positive control drug. Carmichaedine (76) [321] displayed potent antibacterial activity against Bacillus subtilis with a minimum inhibitory concentration of 8 µg/mL. Spicatine A (77) [322] suppressed ConA-induced or LPS-induced splenocyte proliferation in a concentration-dependent manner, with IC50 values of 2.644 ± 0.77 and 2.283 ± 1.28 µmol/L, respectively. 6β-Methoxy,9β-dihydroxylheteratisine (78), heteratisine (79), and 1α,11,13β-trihydroxylhetisine (80) [323] showed significant antioxidant and anti-cholinesterase activities stronger than the positive control drug. Songorine (81) [324] exhibited significant disaggregation potency on the Aβ1−42 aggregates. 8-O-Methyllycaconitine (82) and acosinomonine B (83) [325] showed a potent inhibitory effect on the capsaicin (selective TRPV1 agonist) mediated activation of TRPV1 channels expressed in HEK293 cells with inhibition rates of 30.94% and 31.78% at the concentration of 10 µmol/L. Shawurensine (84) [326] displayed considerably potent antifeedant activity (EC50 = 0.42 and 0.81 mg/cm2 in the choice test and no choice test, respectively). Cammaconine (85) [327] exhibited substantial cardiotonic activity. Notably, a large number of studies indicated that DAs possibly caused disordered ion channels and DNA damage, resulting in mitochondrial-induced cardiomyocyte apoptosis [328].

|
Download:
|
| Fig. 17. The structure of representative bioactive diterpenoid alkaloids over the past decade. | |
{kind=link}
This review summarizes naturally occurring DAs reported from 2013 to 2023 and points out the candidates for leading compounds (Fig. 17). DAs displayed significant protective effects on the cardiovascular system, nervous system, and immune system as well as anti-tumor activity. However, due to a narrow therapeutic window, DAs easily trigger strong cardiotoxicity, neurotoxicity, and liver toxicity, which restrict their practical use. Thus, in-depth research on DAs and their pharmacokinetics and potential mechanisms is conducive to improving the safety of clinical medications, giving full play to analgesia, anti-inflammatory, anti-arrhythmic, anti-tumor, and other effects.
Undoubtedly, more and more DAs with significant biological activities will be discovered in the future with the development of modern analytical, chemical, and molecular biological research techniques. Semi-synthetic variants of DAs are also an alternative source of DAs with lower toxicity and better pharmaceutical activities [329-331]. Furthermore, discovering new biological activities from reported compounds is also a reliable approach to a wider therapeutic window.
Acknowledgment: This work was financially supported by grants from the National Natural Science Foundation of China (No. 82073734).
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statementYingjie Wang: Writing – original draft, Validation, Resources, Investigation, Data curation. Peng Tang: Writing – original draft, Methodology, Investigation, Data curation. Wenchao Tu: Writing – original draft, Methodology, Investigation, Data curation. Qi Gao: Writing – original draft, Visualization, Investigation, Data curation. Cuizhu Wang: Writing – original draft, Project administration, Investigation, Data curation. Luying Tan: Writing – original draft, Investigation, Data curation. Lixin Zhao: Writing – original draft, Methodology, Investigation, Formal analysis. Hongye Han: Writing – original draft, Visualization, Investigation, Data curation. Liefeng Ma: Writing – original draft, Validation, Methodology, Investigation. Kouharu Otsuki: Writing – original draft, Project administration, Investigation, Data curation. Weilie Xiao: Writing – review & editing, Funding acquisition, Data curation, Conceptualization. Wenli Wang: Writing – review & editing, Supervision, Formal analysis, Data curation. Jinping Liu: Writing – review & editing, Validation, Funding acquisition, Formal analysis. Yong Li: Writing – review & editing, Funding acquisition, Formal analysis, Data curation. Zhajun Zhan: Writing – review & editing, Project administration, Formal analysis, Conceptualization. Wei Li: Writing – review & editing, Resources, Project administration, Formal analysis. Xianli Zhou: Writing – review & editing, Supervision, Funding acquisition, Formal analysis. Ning Li: Writing – review & editing, Supervision, Funding acquisition, Formal analysis, Conceptualization.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109955.
| [1] |
D.J. Newman, G.M. Cragg, J. Nat. Prod. 83 (2020) 770-803. DOI:10.1021/acs.jnatprod.9b01285 |
| [2] |
Z.W. Lou, F.C. Yin, X.B. Wang, L.Y. Kong, Chin. J. Nat. Med. 22 (2024) 195-211. |
| [3] |
R.H. Xie, L.S. Li, X.N. Fan, et al., Chin. Chem. Lett. 31 (2020) 431-433. DOI:10.1016/j.cclet.2019.07.068 |
| [4] |
J.B. Gao, X.J. Zhang, K. Shang, et al., Chin. Chem. Lett. 31 (2020) 427-430. DOI:10.1016/j.cclet.2019.09.020 |
| [5] |
Z.J. Zhan, S. Li, W. Chu, S. Yin, Nat. Prod. Rep. 39 (2022) 2132-2174. DOI:10.1039/d2np00047d |
| [6] |
Y. Liu, Z. Song, Y.J. Liu, et al., Acta Pharm. Sin. B 11 (2021) 1513-1525. DOI:10.1016/j.apsb.2021.05.006 |
| [7] |
Y.Y. Feng, S.Q. Zha, H.Q. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107742. DOI:10.1016/j.cclet.2022.107742 |
| [8] |
Y. Li, Y.X. Zhu, Z.X. Zhang, et al., Acta Pharm. Sin. B 10 (2020) 1073-1082. DOI:10.1016/j.apsb.2019.10.013 |
| [9] |
M. Zhang, K. Otsuki, T. Kikuchi, et al., J. Nat. Med. 84 (2021) 2366-2373. DOI:10.1021/acs.jnatprod.1c00570 |
| [10] |
Y.L. Wu, Y.Y. Yang, W. Wang, et al., Acta Pharm. Sin. B 12 (2022) 4262-4265. DOI:10.1016/j.apsb.2022.07.007 |
| [11] |
W.Y. Zhou, Z.H. Xi, N.N. Du, et al., Chin. Chem. Lett. 35 (2024) 109030. DOI:10.1016/j.cclet.2023.109030 |
| [12] |
J.G. Song, J. Zheng, R.J. Wei, et al., Chem 10 (2024) 924-937. DOI:10.1016/j.chempr.2023.12.004 |
| [13] |
A.C.S. Pais, J.A. Saraiva, S.M. Rocha, et al., Mar. Drugs. 17 (2019) 556. DOI:10.3390/md17100556 |
| [14] |
J. Muñoz, G. Culioli, M. Köck, Phytochem. Rev. 12 (2013) 407-424. DOI:10.1007/s11101-012-9246-4 |
| [15] |
A. Arciniegas, A. Pérez-Castorena, K. González, et al., J. Brazil. Chem. Soc. 24 (2013) 1167-1171. |
| [16] |
P.P. Zhang, L.T. Cui, Z.R. Cui, et al., Fitoterapia 159 (2022) 105192. DOI:10.1016/j.fitote.2022.105192 |
| [17] |
S. Xue, P.P. Zhang, P.F. Tang, et al., Fitoterapia 142 (2020) 104518. DOI:10.1016/j.fitote.2020.104518 |
| [18] |
X.P. Zhang, Y.F. Tan, Y.B. Li, et al., Chem. Pharm. Bull. 62 (2014) 494-498. DOI:10.1248/cpb.c14-00056 |
| [19] |
R. Zhang, H.P. He, Y.T. Di, et al., Fitoterapia 92 (2014) 100-104. DOI:10.1016/j.fitote.2013.10.014 |
| [20] |
H.Y. Zhang, C.M. Yuan, M.M. Cao, et al., Phytochem. Lett. 8 (2014) 81-85. DOI:10.1016/j.phytol.2014.02.005 |
| [21] |
H.J. Zhang, J. Luo, S.M. Shan, et al., Org. Lett. 15 (2013) 5512-5515. DOI:10.1021/ol402516p |
| [22] |
D.T. Trang, P.T.T. Huong, N.T. Cuc, et al., Nat. Prod. Commun. 16 (2021) 569-587. |
| [23] |
W.H. Zhong, M. Li, R. Gu, et al., Chem. Biodivers. 19 (2022) e202200898. DOI:10.1002/cbdv.202200898 |
| [24] |
C. Premprasert, S. Tewtrakul, A. Plubrukarn, et al., J. Nat. Med. 67 (2012) 174-181. |
| [25] |
Y.J. Wang, G. Chen, Q.Q. Meng, et al., Phytochemistry 194 (2022) 113019. DOI:10.1016/j.phytochem.2021.113019 |
| [26] |
S. Vangelis, M. Christian, K. Marcel, et al., Mar. Drugs 15 (2017) 245. DOI:10.3390/md15080245 |
| [27] |
V. Smyrniotopoulos, C. Merten, D. Firsova, et al., Mar. Drugs 18 (2020) 581. DOI:10.3390/md18110581 |
| [28] |
V. Smyrniotopoulos, D. Firsova, H. Fearnhead, et al., Mar. Drugs 19 (2021) 42. DOI:10.3390/md19010042 |
| [29] |
D.L.R. Carolina, M.J. Ortega, H. Zbakh, et al., J. Nat. Prod. 79 (2016) 395-405. DOI:10.1021/acs.jnatprod.5b01067 |
| [30] |
M.T. Islam, E.S. Ali, S.J. Uddin, et al., Food Chem. Toxicol. 121 (2018) 82-94. DOI:10.1016/j.fct.2018.08.032 |
| [31] |
M.T. Islam, M.V. de Alencar, K. da Conceição Machado, et al., Chem. Biol. Interact. 240 (2015) 60-73. DOI:10.5958/2229-4503.2015.00005.3 |
| [32] |
W.M. Aung, S. Songkro, S. Songkharak, et al., Saudi Pharm. J. 30 (2022) 679-692. DOI:10.1016/j.jsps.2022.04.004 |
| [33] |
M. Sasaki, T. Mizoshita, T. Mizushima, et al., J. Antimicrob. Chemother. 60 (2007) 1060-1063. DOI:10.1093/jac/dkm329 |
| [34] |
C. Chaotham, W. De-Eknamkul, P. Chanvorachote, J. Nat. Med. 67 (2013) 311-319. DOI:10.1007/s11418-012-0683-6 |
| [35] |
X.Y. Qin, H. Suzuki, M. Honda, et al., Proc. Natl. Acad. Sci. U. S. A. 115 (2018) 4969-4974. DOI:10.1073/pnas.1802279115 |
| [36] |
M. Funaki, J. Kitabayashi, T. Shimakami, et al., Sci. Rep. 7 (2017) 16978. DOI:10.1038/s41598-017-17285-2 |
| [37] |
H.B. Guan, Y.Z. Nie, Y.W. Zheng, et al., Stem Cell Res. Ther. 6 (2015) 51. DOI:10.1186/s13287-015-0046-9 |
| [38] |
H. Okada, R. Takabatake, M. Honda, et al., Oncotarget 8 (2017) 39978-39993. DOI:10.18632/oncotarget.18116 |
| [39] |
K. Okita, N. Izumi, K. Ikeda, et al., J. Gastroenterol. 50 (2015) 667-674. DOI:10.1007/s00535-014-0996-1 |
| [40] |
S. Nakanishi, K. Kinoshita, Y. Kurauchi, et al., Eur. J. Pharmacol. 954 (2023) 175899. DOI:10.1016/j.ejphar.2023.175899 |
| [41] |
N.T. Tam, T.D. Quan, D.V. Hau, et al., Nat. Prod. Res. 33 (2019) 3357-3363. DOI:10.1080/14786419.2018.1477149 |
| [42] |
S.R.M. Ibrahim, G.A.A. Mohamed, Chin. J. Nat. Med. 15 (2017) 546-549. |
| [43] |
X. Gao, J. He, X.D. Wu, et al., Fitoterapia 116 (2017) 116-120. DOI:10.1016/j.fitote.2016.11.018 |
| [44] |
J. Xu, M. Wang, Z. Liu, et al., J. Nat. Prod. 86 (2023) 330-339. DOI:10.1021/acs.jnatprod.2c00937 |
| [45] |
A. Paichitrojjana, A. Paichitrojjana, Drug Des. Devel. Ther. 17 (2023) 2573-2591. DOI:10.2147/dddt.s427530 |
| [46] |
L. Zhang, Y. Yang, B. Wang, et al., J. Am. Acad. Dermatol. 89 (2023) 1141-1148. DOI:10.1016/j.jaad.2023.07.1023 |
| [47] |
A. King, M.G. Tan, C. Kirshen, et al., J. Am. Acad. Dermatol. 89 (2023) 1063-1066. DOI:10.1016/j.jaad.2023.07.010 |
| [48] |
J.S. Barbieri, JAMA Dermatol 159 (2023) 1403. DOI:10.1001/jamadermatol.2023.2065 |
| [49] |
X.H. Su, W.P. Li, Y.J. Wang, et al., Neurotherapeutics 19 (2022) 421-433. DOI:10.1007/s13311-021-01168-6 |
| [50] |
E. Mahase, BMJ 383 (2023) 2545. |
| [51] |
K. Kridin, R.J. Ludwig, J. Am. Acad. Dermatol. 89 (2023) e251-e252. DOI:10.1016/j.jaad.2023.04.049 |
| [52] |
M.C. Dispenza, E.B. Wolpert, K.L. Gilliland, et al., J. Invest. Dermatol. 132 (2012) 2198-2205. DOI:10.1038/jid.2012.111 |
| [53] |
F. Recchia, S. De Filippis, M. Rosselli, et al., Clin. Cancer Res. 7 (2001) 1251-1257. |
| [54] |
M. Jakaria, A. Belaidi, A. Bush, et al., Biomed. Pharmacother. 164 (2023) 114930. DOI:10.1016/j.biopha.2023.114930 |
| [55] |
L. Kousalya, N. Madhav, O. Zianne, et al., Front. Cell. Dev. Biol. 11 (2023) 1254612. DOI:10.3389/fcell.2023.1254612 |
| [56] |
N. Jan, S. Sofi, H. Qayoom, et al., Crit. Rev. Oncol. Hematol. 192 (2023) 104156. DOI:10.1016/j.critrevonc.2023.104156 |
| [57] |
S. Priyamvada, A.N. Anbazhagan, A. Kumar, et al., Inflamm. Bowel. Dis. 26 (2020) 534-545. DOI:10.1093/ibd/izz249 |
| [58] |
G. Valeria, H. Nadia, G. Eugenia, et al., Pharmaceutics 12 (2020) 707. DOI:10.3390/pharmaceutics12080707 |
| [59] |
B. Ramchatesingh, V.A. Martinez, D. Arcuri, et al., Int. J. Mol. Sci. 23 (2022) 12622. DOI:10.3390/ijms232012622 |
| [60] |
H.Y. Yang, M. Liu, Y. Sheng, et al., Acta Pharmacol. Sin. 43 (2022) 441-1452. DOI:10.3390/aerospace9080441 |
| [61] |
H.K. Lee, H.Y. Yoon, K.L. Jang, Biochem. Biophys. Res. Commun. 594 (2022) 15-21. DOI:10.1016/j.bbrc.2022.01.052 |
| [62] |
L.Q. Tong, L. Wang, S. Liao, MBio 13 (2022) e0148522. DOI:10.1128/mbio.01485-22 |
| [63] |
E.S. Pistoia, T. Cosio, E. Campione, J. Fungi 8 (2022) 1049. DOI:10.3390/jof8101049 |
| [64] |
Q. Deng, J.X. Chen, Biomolecules 12 (2022) 869. DOI:10.3390/biom12070869 |
| [65] |
L.L. Tao, Y.M. Nie, G.X. Wang, Mol. Med. Rep. 17 (2018) 2619-2625. |
| [66] |
T.A. Trinh, T.X. Hoang, J.Y. Kim, Mol. Cell. Biochem. 473 (2020) 167-177. DOI:10.1007/s11010-020-03817-4 |
| [67] |
A.F. Khafaga, Y.S. El-Sayed, Naunyn Schmiedebergs Arch. Pharmacol. 391 (2018) 59-70. DOI:10.1007/s00210-017-1437-5 |
| [68] |
H. Shimizu, T. Tsubota, K. Kanki, et al., J. Cell. Physiol. 233 (2018) 607-616. DOI:10.1002/jcp.25921 |
| [69] |
M.G. Ewees, T.M. Abdelghany, A.H. Abdel, et al., Naunyn Schmiedebergs Arch. Pharmacol. 388 (2015) 931-938. DOI:10.1007/s00210-015-1130-5 |
| [70] |
B. Petersen, G.B. Drug Des, Devel. Ther. 3 (2009) 51-57. |
| [71] |
C. Cheng, J. Michaels, N. Scheinfeld, Expert. Opin. Investig. Drugs 17 (2008) 437-443. DOI:10.1517/13543784.17.3.437 |
| [72] |
R. Rosas, S. Buryska, R. Silver, et al., Anticancer Res. 40 (2020) 3071-3080. DOI:10.21873/anticanres.14288 |
| [73] |
H. Yang, Y. Tao, M. Zhang, et al., Anticancer Drugs 30 (2019) 56-64. DOI:10.1097/CAD.0000000000000692 |
| [74] |
Z.Q. Yuan, G.Y. Wang, J.W. Qu, et al., Biochem. Biophys. Res. Commun. 503 (2018) 188-194. DOI:10.1016/j.bbrc.2018.06.001 |
| [75] |
V.M. Manzano, J.C. Muñoz, J.R. Jiménez, et al., Br. J. Pharmacol. 131 (2000) 1673-1683. DOI:10.1038/sj.bjp.0703728 |
| [76] |
B. Kubickova, S. Martinkova, D. Bohaciakova, et al., Toxicology 487 (2023) 153461. DOI:10.1016/j.tox.2023.153461 |
| [77] |
A.M. Mohrbacher, A.S. Yang, S. Groshen, et al., Clin. Cancer Res. 23 (2017) 4550-4555. DOI:10.1158/1078-0432.CCR-17-0234 |
| [78] |
F. Pastorino, C. Brignole, D. Di Paolo, et al., Small 15 (2019) e1804591. DOI:10.1002/smll.201804591 |
| [79] |
L. Galassi, M. Rossi, P. Lodeserto, et al., Pharmaceutics 15 (2023) 648. DOI:10.3390/pharmaceutics15020648 |
| [80] |
S. Pignatta, I. Orienti, M. Falconi, et al., Nanomedicine 11 (2015) 263-273. DOI:10.1016/j.nano.2014.10.004 |
| [81] |
G.C. Salata, I.D. Malagó, V.F.M. Carvalho Dartora, et al., Mol. Pharm. 18 (2021) 3401-3417. DOI:10.1021/acs.molpharmaceut.1c00319 |
| [82] |
X.H. Tang, M. Melis, K. Mai, et al., Front. Pharmacol. 12 (2021) 630557. DOI:10.3389/fphar.2021.630557 |
| [83] |
W.Y. Wang, D.Y. Huang, Y.B. Yu, et al., Angew. Chem. Int. Ed. 62 (2023) e202307626. DOI:10.1002/anie.202307626 |
| [84] |
R. Li, S.L. Morris-Natschke, K.H. Lee, Nat. Prod. Rep. 33 (2016) 1166-1226. DOI:10.1039/C5NP00137D |
| [85] |
D.G. Wirasisya, J. Hohmann, Fitoterapia 166 (2023) 105466. DOI:10.1016/j.fitote.2023.105466 |
| [86] |
N. Zeng, Q.D. Zhang, Q.Y. Yao, et al., Molecules 29 (2024) 60. |
| [87] |
R. Acquaviva, G.A. Malfa, M.R. Loizzo, et al., Molecules 27 (2022) 4791. DOI:10.3390/molecules27154791 |
| [88] |
L. Xie, X.J. Zhang, Y. Wang, et al., Nat. Prod. Res. (2023). DOI:10.1080/14786419.2023.2248541 |
| [89] |
Z. Yu, Y. Meng, M. Xue, et al., Chinese J. Org. Chem. 43 (2023) 2567-2571. DOI:10.6023/cjoc202212020 |
| [90] |
Y.Y. Chen, X.T. Zeng, D.Q. Xu, et al., Phytochemistry 197 (2022) 113113. DOI:10.1016/j.phytochem.2022.113113 |
| [91] |
S.P. Adiguna, J.A. Panggabean, R.T. Swasono, et al., Plants 12 (2023) 1220. DOI:10.3390/plants12061220 |
| [92] |
A. Gour, P. Kotwal, A. Dogra, et al., ACS Omega 7 (2022) 12765-12771. DOI:10.1021/acsomega.1c07339 |
| [93] |
J.K. Tan, R. Chen, R.C.H. Lee, et al., Pharmaceuticals 15 (2022) 115. |
| [94] |
J.P. Loureiro Damasceno, H. Silva da Rosa, L. Silva de Araújo, et al., Eur. J. Drug Metab. Pharmacokinet. 47 (2022) 19-30. DOI:10.1007/s13318-021-00736-7 |
| [95] |
Y. Yan, L.H. Fang, G.H. Du, Andrographolide, in: G.H. Du (Ed.), Natural Small Molecule Drugs from Plants, Springer Singapore, Singapore, 2018.
|
| [96] |
B. Zeng, A. Wei, Q. Zhou, et al., Phytother. Res. 36 (2022) 336-364. DOI:10.1002/ptr.7324 |
| [97] |
S. Wei, Y.B. Tang, H. Hua, et al., Bioorg. Med. Chem. Lett. 23 (2013) 4056-4060. DOI:10.1016/j.bmcl.2013.05.061 |
| [98] |
G. Kumar, D. Singh, J.A. Tali, et al., Bioorg. Chem. 95 (2020) 103511. DOI:10.1016/j.bioorg.2019.103511 |
| [99] |
A. Basu, S. Guti, S. Kundu, et al., J. Drug Deliv. Sci. Tec. 55 (2020) 101406. DOI:10.1016/j.jddst.2019.101406 |
| [100] |
Y. Zhou, M. Yang, X. Yan, et al., ACS Appl. Mater. Interfaces 15 (2023) 36061-36075. DOI:10.1021/acsami.3c09342 |
| [101] |
Z. Feng, J. Cao, Q. Zhang, et al., Chin. Med. 15 (2020) 126. DOI:10.1186/s13020-020-00407-w |
| [102] |
Y. Li, J.M. Li, Y.J. Xu, et al., Fitoterapia 171 (2023) 105654. DOI:10.1016/j.fitote.2023.105654 |
| [103] |
D.W. Bi, Y.X. Zhao, X. Qiu, et al., Chem. Biodivers. 20 (2023) e202200985. DOI:10.1002/cbdv.202200985 |
| [104] |
D.B. Pu, J. Lin, X.J. Pu, et al., Bioorg. Chem. 128 (2022) 106022. DOI:10.1016/j.bioorg.2022.106022 |
| [105] |
A. Ledoux, C. Hamann, O. Bonnet, et al., Molecules 28 (2023) 1197. DOI:10.3390/molecules28031197 |
| [106] |
R. Irshad, A.S.A. Kabbashi, K.M. Salawu, et al., Fitoterapia 160 (2022) 105226. DOI:10.1016/j.fitote.2022.105226 |
| [107] |
Y.L. Zhu, L. Deng, J.Q. Song, et al., Org. Chem. Front. 9 (2022) 6945-6957. DOI:10.1039/d2qo01437h |
| [108] |
R.B. Hernández-Alvarado, A. Madariaga-Mazón, A. Ortega, et al., ACS Chem. Neurosci. 11 (2020) 3979-3992. DOI:10.1021/acschemneuro.0c00608 |
| [109] |
T.E. Prisinzano, R.B. Rothman, Chem. Rev. 108 (2008) 1732-1743. DOI:10.1021/cr0782269 |
| [110] |
J. Xu, W. Chen, Z. Feng, et al., Med. Chem. Res. 31 (2022) 695-704. DOI:10.1007/s00044-022-02868-0 |
| [111] |
X.G. Liu, X. Lu, W. Gao, et al., Nat. Prod. Rep. 39 (2022) 474-511. DOI:10.1039/d1np00026h |
| [112] |
P. Tang, T. Shen, H. Wang, et al., Biomed. Pharmacother. 164 (2023) 114955. DOI:10.1016/j.biopha.2023.114955 |
| [113] |
D. Halder, S. Das, et al., J. Biomol. Struct. Dyn. 41 (2023) 11286-11323. DOI:10.1080/07391102.2022.2158136 |
| [114] |
M. Gaborova, K. Smejkal, R. Kubinova, Molecules 27 (2022) 166. |
| [115] |
H. Bao, Q. Zhang, Y. Ye, et al., Phytochem. Rev. 16 (2017) 235-270. DOI:10.1007/s11101-016-9472-2 |
| [116] |
R.J. Grayer, A.J. Paton, M.S.J. Simmonds, et al., Nat. Prod. Rep. 38 (2021) 1720-1728. DOI:10.1039/d0np00081g |
| [117] |
M.A. Gonzalez, Nat. Prod. Rep. 32 (2015) 684-704. DOI:10.1039/C4NP00110A |
| [118] |
W.H. Lin, J.M. Fang, Y.S. Cheng, Phytochemistry 46 (1997) 169-173. DOI:10.1016/S0031-9422(97)00267-7 |
| [119] |
W.H. Lin, J.M. Fang, Y.S. Cheng, Phytochemistry 42 (1996) 1657-1663. DOI:10.1016/0031-9422(96)00198-7 |
| [120] |
J.R. Hanson, Nat. Prod. Rep. 34 (2017) 1233-1243. DOI:10.1039/C7NP00040E |
| [121] |
J.J. Chen, H.M. Wu, C.F. Peng, et al., J. Nat. Prod. 72 (2009) 223-228. DOI:10.1021/np800721f |
| [122] |
W.F. Chen, L.L. Wong, X. Zhang, et al., Chem. Biodivers. 16 (2019) e1900206. DOI:10.1002/cbdv.201900206 |
| [123] |
H. Van Oanh, P.X. Sinh, N.T. An, et al., Nat. Prod. Commun. 4 (2009) 323-325. |
| [124] |
A. Shakeri, S.S. Farahmand, Z. Tayarani-Najaran, et al., Naunyn Schmiedebergs Arch. Pharmacol. 394 (2021) 241-248. DOI:10.1007/s00210-020-01967-2 |
| [125] |
G.F. Lai, X.Y. Wang, Y.F. Wang, et al., Helvet. Chim. Acta 92 (2009) 470-480. DOI:10.1002/hlca.200800274 |
| [126] |
D.B. Pu, T. Wang, X.J. Zhang, et al., RSC Adv. 8 (2018) 6425-6435. DOI:10.1039/c7ra13309j |
| [127] |
Z.H. Wang, C. Niu, D.J. Zhou, et al., Molecules 22 (2017). |
| [128] |
X.L. Yan, J.S. Zhang, J.L. Huang, et al., Phytochemistry 166 (2019) 112064. DOI:10.1016/j.phytochem.2019.112064 |
| [129] |
M. Li, F. He, Y. Zhou, et al., Arch. Pharm. Res. 42 (2019) 512-518. DOI:10.1007/s12272-019-01151-y |
| [130] |
J.C. Li, Z.J. Zhang, T. Yang, et al., Phytochem. Lett. 34 (2019) 13-17. DOI:10.1016/j.phytol.2019.09.003 |
| [131] |
L. Chen, Q. Yang, K. Hu, et al., Fitoterapia 134 (2019) 158-164. DOI:10.24963/ijcai.2019/23 |
| [132] |
Z.P. Jiang, L.W. Tian, L. Shen, et al., Fitoterapia 130 (2018) 272-280. DOI:10.1016/j.fitote.2018.09.011 |
| [133] |
Y.D. Zhu, J.Y. Zhang, P.F. Li, et al., Nat. Prod. Res. 30 (2016) 1075-1080. DOI:10.1080/14786419.2015.1107061 |
| [134] |
Y.F. Wang, H.B. Fang, Y.M. Yan, et al., Phytochemistry 215 (2023) 113835. DOI:10.1016/j.phytochem.2023.113835 |
| [135] |
X. Liu, X.Q. Zhan, M.J. Wang, Phytochemistry 205 (2023) 113501. DOI:10.1016/j.phytochem.2022.113501 |
| [136] |
R. Li, X.R. Sun, X.T. He, et al., Fitoterapia 168 (2023) 105519. DOI:10.1016/j.fitote.2023.105519 |
| [137] |
G.J. Kim, E.J. Yang, Y.S. Kim, et al., Phytochemistry 211 (2023) 113711. DOI:10.1016/j.phytochem.2023.113711 |
| [138] |
X. Fu, D. Yu, G. Zhu, et al., Phytochem. Lett. 55 (2023) 56-60. DOI:10.1016/j.phytol.2023.02.009 |
| [139] |
Q.F. Zhu, G.B. Xu, S.G. Liao, et al., Molecules 27 (2022) 7258. DOI:10.3390/molecules27217258 |
| [140] |
J. Li, X. Meng, C. Yin, et al., Chin. J. Nat. Med. 21 (2023) 619-630. DOI:10.56028/aehssr.7.1.619.2023 |
| [141] |
Y.J. Hu, Q. Lan, B.J. Su, et al., Phytochemistry 201 (2022) 113255. DOI:10.1016/j.phytochem.2022.113255 |
| [142] |
Z. Alizadeh, G. Donadio, M.M. Farimani, et al., Phytochemistry 191 (2021) 112926. DOI:10.1016/j.phytochem.2021.112926 |
| [143] |
L. Yang, P. Guo, A. Liu, et al., Fitoterapia 133 (2019) 180-185. DOI:10.1109/arso46408.2019.8948763 |
| [144] |
T. Murata, Y. Ishikawa, E. Saruul, et al., Phytochemistry 130 (2016) 152-158. DOI:10.1016/j.phytochem.2016.05.011 |
| [145] |
S. Xu, P. Liu, Expert Opin. Ther. Pat. 23 (2013) 149-153. DOI:10.1517/13543776.2013.743995 |
| [146] |
J. Fang, P.J. Little, S. Xu, Med. Res. Rev. 38 (2018) 201-228. DOI:10.1002/med.21438 |
| [147] |
X. Zhao, B. Cacherat, Q. Hu, D. Ma, Nat. Prod. Rep. 39 (2022) 119-138. DOI:10.1039/d1np00028d |
| [148] |
W.C. Tu, Y.X. Huang, B. Li, et al., J. Nat. Prod. 10 (2023) 2348-2359. DOI:10.1021/acs.jnatprod.3c00543 |
| [149] |
M. Zhang, C. Zhao, W.F. Dai, et al., Phytochemistry 137 (2017) 174-181. DOI:10.1016/j.phytochem.2017.02.021 |
| [150] |
D. Li, T. Han, S. Xu, et al., Molecules 21 (2016) 575. DOI:10.3390/molecules21050575 |
| [151] |
J. Zhu, Y. Sun, Y. Lu, et al., Cell Death Dis. 9 (2018) 708. DOI:10.1038/s41419-018-0684-9 |
| [152] |
M.S. Nogueira, F.B. Da Costa, R. Brun, et al., Molecules 21 (2016) 1237. DOI:10.3390/molecules21091237 |
| [153] |
Y. Liu, X. Wu, J. An, et al., J. Cell Biochem. 120 (2019) 6137-6144. DOI:10.1002/jcb.27901 |
| [154] |
J.H. Huang, C.C. Lan, Y.T. Hsu, et al., Evid. Based Complement. Alternat. Med. 2020 (2020) 9724520. DOI:10.1155/2020/9724520 |
| [155] |
Y.J. Zhao, H. Lv, P.B. Xu, et al., Kaohsiung J. Med. Sci. 32 (2016) 452-457. DOI:10.1016/j.kjms.2016.07.013 |
| [156] |
K.H. Lin, C.Y. Li, Y.M. Hsu, et al., Food Chem. Toxicol. 133 (2019) 110765. DOI:10.1016/j.fct.2019.110765 |
| [157] |
C. Lu, C. Chen, A. Chen, et al., Evid. Based Complement. Alternat. Med. 2020 (2020) 7395187. DOI:10.1155/2020/7395187 |
| [158] |
K.H. Zang, Y.Y. Shao, X. Zuo, et al., J. Med. Food. 19 (2016) 586-592. DOI:10.1089/jmf.2015.3595 |
| [159] |
W. Guo, P. Zheng, J. Zhang, et al., Int. Immunopharmacol. 17 (2013) 1148-1154. DOI:10.1016/j.intimp.2013.10.023 |
| [160] |
D. Liu, H. Qin, B. Yang, et al., Drug Dev. Res. 81 (2020) 526-533. DOI:10.1002/ddr.21649 |
| [161] |
P.J.M. Sobral, A.T.S. Vicente, J.A.R. Salvador, Front. Chem. 11 (2023) 1066280. DOI:10.3389/fchem.2023.1066280 |
| [162] |
Y. Ding, C. Ding, N. Ye, et al., Eur. J. Med. Chem. 122 (2016) 102-117. DOI:10.1016/j.ejmech.2016.06.015 |
| [163] |
S. Li, D. Shi, L. Zhang, et al., Exp. Ther. Med. 16 (2018) 4859-4864. |
| [164] |
D. Luo, Y. Yi, K. Peng, et al., Eur. J. Med. Chem. 178 (2019) 365-379. DOI:10.1016/j.ejmech.2019.06.006 |
| [165] |
D.L. Liu, H.Q. Bu, W.L. Wang, et al., Transl. Cancer Res. 9 (2020) 4148-4161. DOI:10.21037/tcr-19-3000 |
| [166] |
Y.L. Dong, C.P. Huang, J. Li, Acta Med. Mediterr. 34 (2018) 819-825. |
| [167] |
Z. He, X. Xiao, S. Li, et al., Oncol. Lett. 14 (2017) 2499-2504. DOI:10.3892/ol.2017.6421 |
| [168] |
K. Chen, J. Ye, L. Qi, et al., Anticancer Drugs 30 (2019) 925-932. DOI:10.1097/cad.0000000000000797 |
| [169] |
I.H. Yang, J.A. Shin, K.E. Lee, et al., Eur. J. Oral Sci. 125 (2017) 438-443. DOI:10.1111/eos.12387 |
| [170] |
W. Liu, G. Huang, Y. Yang, et al., Int. J. Med. Sci. 18 (2021) 81-87. DOI:10.7150/ijms.48552 |
| [171] |
O. Kadioglu, M. Saeed, V. Kuete, et al., Front. Pharmacol. 9 (2018) 355. DOI:10.3389/fphar.2018.00355 |
| [172] |
H. Wu, G.H. Zhu, Y.Z. Su, Nutraceutical Res. 18 (2020) 292-296. |
| [173] |
Y. Lu, Y. Sun, J. Zhu, et al., Cell Death Dis. 9 (2018) 15. DOI:10.1038/s41419-017-0031-6 |
| [174] |
W. Li, L. Ma, J. Cell Biochem. 120 (2019) 5620-5627. DOI:10.1002/jcb.27845 |
| [175] |
J. Lu, X. Chen, S. Qu, et al., Oncol. Lett. 13 (2017) 2838-2846. DOI:10.3892/ol.2017.5751 |
| [176] |
J.H. Jiang, J. Pi, H. Jin, et al., J. Cell Biochem. 120 (2019) 3736-3746. DOI:10.1002/jcb.27654 |
| [177] |
D. Zhang, Q. Zhou, D. Huang, et al., Biochem. Biophys. Res. Commun. 513 (2019) 594-601. DOI:10.1016/j.bbrc.2019.04.011 |
| [178] |
C. Li, Q. Wang, S. Shen, et al., Oncol. Lett. 16 (2018) 2289-2298. DOI:10.3390/app8112289 |
| [179] |
J. Liu, N. Zhang, N. Li, et al., Pharm. Biol. 57 (2019) 787-791. DOI:10.1080/13880209.2019.1688844 |
| [180] |
W. Xu, J. Sun, T.T. Zhang, et al., Acta Pharmacol. Sin. 27 (2006) 1642-1646. DOI:10.1111/j.1745-7254.2006.00440.x |
| [181] |
X. Li, C.T. Zhang, W. Ma, et al., Front. Pharmacol. 12 (2021) 645824. DOI:10.3389/fphar.2021.645824 |
| [182] |
Q.Q. Liu, H.L. Wang, K. Chen, et al., J. Dig. Dis. 17 (2016) 104-112. DOI:10.1111/1751-2980.12314 |
| [183] |
M. Liu, D.R. Littler, J. Rossjohn, et al., ACS Omega 7 (2022) 7327-7332. DOI:10.1021/acsomega.1c07186 |
| [184] |
X. Hu, Z. Bai, J. Qiao, et al., Eur. J. Med. Chem. 171 (2019) 169-179. DOI:10.1016/j.ejmech.2019.03.046 |
| [185] |
K.H. Kim, R.T. Sadikot, M. Joo, Biochem. Biophys. Res. Commun. 474 (2016) 534-540. DOI:10.1016/j.bbrc.2016.04.122 |
| [186] |
D.P. Dalenogare, P.R. Ferro, S.D.T. De Prá, et al., Inflammopharmacology 27 (2019) 829-844. DOI:10.1007/s10787-019-00588-3 |
| [187] |
C.H. Martins, F. Abrão, T.S. Moraes, et al., Future Microbiol. 13 (2018) 1585-1601. DOI:10.2217/fmb-2018-0140 |
| [188] |
Â.V.F. Alves, C.R. Melo, J.L. Chagas-Neto, et al., Int. J. Pharm. 631 (2023) 122497. DOI:10.1016/j.ijpharm.2022.122497 |
| [189] |
L. Marcondes-Alves, V. Fattori, S.M. Borghi, et al., Nat. Prod. Res. 33 (2019) 921-924. DOI:10.1080/14786419.2017.1416372 |
| [190] |
D. Kian, C.A.C. Lancheros, J.P. Assolini, et al., Biomed. Pharmacother. 103 (2018) 1294-1301. DOI:10.1016/j.biopha.2018.04.164 |
| [191] |
T.C. Okoye, P.A. Akah, E.O. Omeje, et al., Pharmacol. Biochem. Behav. 109 (2013) 38-43. DOI:10.1016/j.pbb.2013.05.001 |
| [192] |
C.W. Han, J. Korean Med. 4 (2020) 64-71. |
| [193] |
P.M. Matos, B. Mahoney, Y. Chan, et al., Molecules 20 (2015) 18264-18278. DOI:10.3390/molecules201018264 |
| [194] |
X. Deng, Y. Shen, J. Yang, et al., Eur. J. Med. Chem. 65 (2013) 403-414. DOI:10.1016/j.ejmech.2013.05.010 |
| [195] |
S.M. Borghi, T.P. Domiciano, F.S. Rasquel-Oliveira, et al., J. Ethnopharmacol. 283 (2022) 114708. DOI:10.1016/j.jep.2021.114708 |
| [196] |
D.M. Matos, M.R. Viana, M.C.O. Alvim, et al., Fitoterapia 128 (2018) 142-147. DOI:10.1016/j.fitote.2018.05.013 |
| [197] |
N. Osafo, D.D. Obiri, A.O. Antwi, et al., J. Basic Clin. Physiol. Pharmacol. 29 (2018) 659-669. DOI:10.1515/jbcpp-2018-0019 |
| [198] |
R.P. Biney, C.K. Benneh, J.O. Kyekyeku, et al., Pharmaceut. Biosci. J. 6 (2018) 7-16. |
| [199] |
R.P. Biney, C.K. Benneh, D.W. Adongo, et al., Psychopharmacology 238 (2021) 2105-2120. DOI:10.1007/s00213-021-05835-6 |
| [200] |
E. Woode, E.O. Ameyaw, E. Boakye-Gyasi, et al., Pharm. Biol. 54 (2016) 2978-2986. DOI:10.1080/13880209.2016.1199040 |
| [201] |
N. Osafo, A.O. Antwi, P.K. Mante, et al., Adv. Tradit. Med. 22 (2022) 649-658. DOI:10.1007/s13596-021-00595-2 |
| [202] |
D. Soh, N. Ernestine, E.M. Tchana Satchet, et al., Nat. Prod. Res. 36 (2022) 1288-1295. DOI:10.1080/14786419.2021.1876045 |
| [203] |
S.A. Osei, R.P. Biney, E. Obese, et al., Malar J 20 (2021) 113. DOI:10.1186/s12936-021-03658-6 |
| [204] |
Y.D. Boakye, N. Osafo, J. Oppong-Kyekyeku, et al., Inflammopharmacology 30 (2022) 1835-1841. DOI:10.1007/s10787-022-00950-y |
| [205] |
J.O. Kyekyeku, S. Asare-Nkansah, S.O. Bekoe, et al., Phytochem. Anal. 31 (2020) 349-354. DOI:10.1002/pca.2901 |
| [206] |
R.N. Alolga, S.L. Wang, I. Ayensu, et al., J. Pharm. Biomed. Anal. 224 (2023) 115200. DOI:10.1016/j.jpba.2022.115200 |
| [207] |
J. Zhou, Z. Zuo, J. Liu, et al., Org. Chem. Front. 7 (2020) 820-828. DOI:10.1039/c9qo01538h |
| [208] |
Y. Li, Y. Liu, H. Yan, et al., Sci. Rep. 6 (2016) 36752. DOI:10.1038/srep36752 |
| [209] |
Y. Li, Y. Liu, J. Zhang, et al., J. Nat. Prod. 78 (2015) 2887-2895. DOI:10.1021/acs.jnatprod.5b00456 |
| [210] |
Y. Li, Y. Liu, J. Zhang, et al., Org. Lett. 15 (2013) 3074-3077. DOI:10.1021/ol401254e |
| [211] |
J. Zhou, N. Sun, H. Zhang, et al., Org. Lett. 19 (2017) 5352-5355. DOI:10.1021/acs.orglett.7b02633 |
| [212] |
Y. Li, Y. Zhu, Z. Zhang, et al., Acta. Pharm. Sin. B 10 (2020) 1073-1082. DOI:10.1016/j.apsb.2019.10.013 |
| [213] |
J. Zhou, G. Zhan, H. Zhang, et al., Org. Lett. 19 (2017) 3935-3938. DOI:10.1021/acs.orglett.7b01863 |
| [214] |
J. Zhou, J. Liu, T. Dang, et al., Org. Lett. 20 (2018) 2063-2066. DOI:10.1021/acs.orglett.8b00606 |
| [215] |
K. Chen, T. Wang, Y. Li, et al., Acta. Pharm. Sin. B 13 (2023) 1326-1336. DOI:10.1016/j.apsb.2023.01.021 |
| [216] |
G. Appendino, Ingenane diterpenoids, in: A. Kinghorn, H. Falk, S. Gibbons, J. Kobayashi (Eds.), Progress in the Chemistry of Organic Natural Products, Springer, Cham, 2016, pp. 1–90.
|
| [217] |
H. Zhao, L. Sun, C. Kong, et al., J. Ethnopharmacol. 298 (2022) 115574. DOI:10.1016/j.jep.2022.115574 |
| [218] |
X.L. Yan, J. Sang, S.X. Chen, et al., Org. Lett. 21 (2019) 4128-4131. DOI:10.1021/acs.orglett.9b01315 |
| [219] |
L.S. Wan, Y. Nian, X.R. Peng, et al., Org. Lett. 20 (2018) 3074-3078. DOI:10.1021/acs.orglett.8b01114 |
| [220] |
F.Y. Yuan, Y.H. Pan, A.P. Yin, et al., Org. Chem. Front. 9 (2022) 775-780. DOI:10.1039/d1qo01705e |
| [221] |
D.Q. Fei, L.L. Dong, F.M. Qi, et al., Org. Lett. 18 (2016) 2844-2847. DOI:10.1021/acs.orglett.6b01093 |
| [222] |
X.H. Meng, K. Wang, T. Chai, et al., Phytochemistry 172 (2020) 112257. DOI:10.1016/j.phytochem.2020.112257 |
| [223] |
M. Liu, F. Chen, R. Yu, et al., Int. J. Mol. Sci. 17 (2016) 1348. DOI:10.3390/ijms17081348 |
| [224] |
Y. Zhang, J.W. Lou, A. Kang, et al., J. Ethnopharmacol. 249 (2020) 112423. DOI:10.1016/j.jep.2019.112423 |
| [225] |
S. Shaker, J. Sang, X.L. Yan, et al., Phytochemistry 176 (2020) 112395. DOI:10.1016/j.phytochem.2020.112395 |
| [226] |
A. Hasan, D. Tang, D. Nijat, et al., Bioorg. Chem. 117 (2021) 105442. DOI:10.1016/j.bioorg.2021.105442 |
| [227] |
C.S. Huang, S.H. Luo, Y.L. Li, et al., Nat. Prod. Bioprospect. 4 (2014) 91-100. DOI:10.1007/s13659-014-0009-3 |
| [228] |
J.Y. Tsai, D. Rédei, P. Forgo, et al., J. Nat. Prod. 79 (2016) 2658-2666. DOI:10.1021/acs.jnatprod.6b00603 |
| [229] |
L.F. Nothias-Scaglia, C. Pannecouque, F. Renucci, et al., J. Nat. Prod. 78 (2015) 1277-1283. DOI:10.1021/acs.jnatprod.5b00073 |
| [230] |
P. Wang, P. Lu, X. Qu, et al., Sci. Rep. 7 (2017) 9451. DOI:10.1038/s41598-017-07157-0 |
| [231] |
G. Jiang, E.A. Mendes, P. Kaiser, et al., AIDS 28 (2014) 1555-1566. DOI:10.1097/QAD.0000000000000289 |
| [232] |
J.X. Zhao, C.P. Liu, W.Y. Qi, et al., J. Nat. Prod. 77 (2014) 2224-2233. DOI:10.1021/np5004752 |
| [233] |
M. Jiang, Y. Xue, J. Li, et al., J. Inflamm. Res. 14 (2021) 2681-2696. DOI:10.2147/jir.s306846 |
| [234] |
N.J. Collier, F.R. Ali, J.T. Lear, Clin. Pract. 11 (2014) 295-306. DOI:10.2217/cpr.14.13 |
| [235] |
S.M. Ogbourne, P.G. Parsons, Fitoterapia 98 (2014) 36-44. DOI:10.1016/j.fitote.2014.07.002 |
| [236] |
C.G. Parker, C.A. Kuttruff, A. Galmozzi, et al., ACS Cent. Sci. 3 (2017) 1276-1285. DOI:10.1021/acscentsci.7b00420 |
| [237] |
C.A. Stoddart, C.M. Abreu, S.L. Price, et al., PLoS One 9 (2014) e97257. DOI:10.1371/journal.pone.0097257 |
| [238] |
J.L. Sloane, N.L. Benner, K.N. Keenan, et al., Proc. Natl. Acad. Sci. U. S. A. 117 (2020) 10688-10698. DOI:10.1073/pnas.1919408117 |
| [239] |
Q. Liu, W. Li, L. Huang, et al., Eur. J. Med. Chem. 156 (2018) 618-627. DOI:10.1016/j.ejmech.2018.07.020 |
| [240] |
L. Jørgensen, S.J. McKerrall, C.A. Kuttruff, et al., Science 341 (2013) 878-882. DOI:10.1126/science.1241606 |
| [241] |
S.J. McKerrall, L. Jørgensen, C.A. Kuttruff, et al., J. Am. Chem. Soc. 136 (2014) 5799-5810. DOI:10.1021/ja501881p |
| [242] |
M.J. Classen, M.N.A. Böcker, R. Roth, et al., J. Am. Chem. Soc. 143 (2021) 8261-8265. DOI:10.1021/jacs.1c04210 |
| [243] |
Z. Liu, Z.W. Ding, K. Chen, et al., Nat. Prod. Rep. 38 (2021) 1589-1617. DOI:10.1039/d0np00086h |
| [244] |
H.B. Wang, X.Y. Wang, L.P. Liu, et al., Chem. Rev. 115 (2015) 2975-3011. DOI:10.1021/cr200397n |
| [245] |
K. Otsuki, W. Li, J. Nat. Med. 77 (2023) 625-643. DOI:10.1007/s11418-023-01713-x |
| [246] |
J. Hyun, M.H. Park, Y.H. Lee, et al., J. Ethnopharmacol. 278 (2021) 114238. DOI:10.1016/j.jep.2021.114238 |
| [247] |
K.C. Du, X.Y. Yang, J.H. Li, et al., Phytochemistry 177 (2020) 112437. DOI:10.1016/j.phytochem.2020.112437 |
| [248] |
C.J. Wang, Q.L. Yan, Y.F. Ma, et al., J. Nat. Prod. 80 (2017) 1248-1254. DOI:10.1021/acs.jnatprod.6b00786 |
| [249] |
H.J. Lee, M. Zhang, T.P. Doan, et al., Phytochemistry 212 (2023) 113740. DOI:10.1016/j.phytochem.2023.113740 |
| [250] |
F.Y. Yuan, Z.Y. Tang, D. Huang, et al., Bioorg. Chem. 128 (2022) 106103. DOI:10.1016/j.bioorg.2022.106103 |
| [251] |
A.H. Wang, X.G. Tian, Y.L. Cui, et al., Phytochemistry 146 (2018) 82-90. DOI:10.1016/j.phytochem.2017.12.005 |
| [252] |
R.J. Ferreira, G. Spengler, A. Orthaber, et al., Org. Lett. 23 (2021) 274-278. DOI:10.1021/acs.orglett.0c03647 |
| [253] |
L. Wang, J. Yang, L.M. Kong, et al., Org. Lett. 19 (2017) 3911-3914. DOI:10.1021/acs.orglett.7b01813 |
| [254] |
Y.C. Tsai, R.A. Nell, J.E. Buckendorf, et al., Pharmaceuticals 14 (2021) 653. DOI:10.3390/ph14070653 |
| [255] |
J.F. Wang, L. Qin, B.Q. Zhao, et al., Org. Biomol. Chem. 17 (2019) 195-202. DOI:10.1039/C8OB02519C |
| [256] |
F. Olivon, H. Palenzuela, E. Girard-Valenciennes, et al., J. Nat. Prod. 78 (2015) 1119-1128. DOI:10.1021/acs.jnatprod.5b00116 |
| [257] |
M. Esposito, L.F. Nothias, P. Retailleau, et al., J. Nat. Prod. 80 (2017) 2051-2059. DOI:10.1021/acs.jnatprod.7b00233 |
| [258] |
N. Geribaldi-Doldán, E. Flores-Giubi, M. Murillo-Carretero, et al., Int. J. Neuropsychopharmacol. 18 (2015) pyv070. DOI:10.1093/ijnp/pyv070 |
| [259] |
S. Chow, T. Krainz, C.J. Bettencourt, et al., Chemistry 26 (2020) 13372-13377. DOI:10.1002/chem.202003221 |
| [260] |
L. Ma, Z. Chen, J. Li, et al., J. Ethnopharmacol. 279 (2021) 113889. DOI:10.1016/j.jep.2021.113889 |
| [261] |
J. Zhang, J. Liu, H. Li, et al., J. Agric. Food Chem. 71 (2023) 12730-12740. DOI:10.1021/acs.jafc.3c03460 |
| [262] |
D. Mochly-Rosen, K. Das, K.V. Grimes, Nat. Rev. Drug Discov. 11 (2012) 937. DOI:10.1038/nrd3871 |
| [263] |
G.K. Brown, J.R. Finlay, R.C. Straw, et al., Front. Vet. Sci. 9 (2022) 1003165. DOI:10.3389/fvets.2022.1003165 |
| [264] |
R.L. Moses, G.M. Boyle, R.A. Howard-Jones, et al., Biochem. Pharmacol. 178 (2020) 114048. DOI:10.1016/j.bcp.2020.114048 |
| [265] |
L.C. Powell, J.K. Cullen, G.M. Boyle, et al., Sci. Transl. Med. 14 (2022) eabn3758. DOI:10.1126/scitranslmed.abn3758 |
| [266] |
H.E. De la Torre-Tarazona, R. Jiménez, P. Bueno, et al., Biochem. Pharmacol. 177 (2020) 113937. DOI:10.1016/j.bcp.2020.113937 |
| [267] |
A.H.H. El-Desoky, K. Eguchi, N. Kishimoto, et al., J. Med. Chem. 65 (2022) 3460-3472. DOI:10.1021/acs.jmedchem.1c01973 |
| [268] |
Y.C. Q. Li, L. Zhao, et al., Med. Chem. Lett. 50 (2021) 128319. DOI:10.1016/j.bmcl.2021.128319 |
| [269] |
A. Ezzanad, R. Gómez-Oliva, F. Escobar-Montaño, et al., J. Med. Chem. 64 (2021) 6070-6084. DOI:10.1021/acs.jmedchem.1c00156 |
| [270] |
Q.Q. Song, Y. Rao, G.H. Tang, et al., J. Med. Chem. 62 (2019) 2060-2075. DOI:10.1021/acs.jmedchem.8b01693 |
| [271] |
A.J. Catino, A. Sherlock, P. Shieh, et al., Org. Lett. 15 (2013) 3330-3333. DOI:10.1021/ol401367h |
| [272] |
G. Tong, Z. Liu, P. Li, et al., Org. Lett. 16 (2014) 2288-2291. DOI:10.1021/ol5008263 |
| [273] |
S. Kawamura, H. Chu, J. Felding, et al., Nature 532 (2016) 90-93. DOI:10.1038/nature17153 |
| [274] |
Y. Li, M. Wei, M. Dai, et al., Tetrahedron 73 (2017) 4172-4177. DOI:10.1016/j.tet.2016.11.005 |
| [275] |
G. Tong, Z. Ding, Z. Liu, et al., J. Org. Chem. 85 (2020) 4813-4837. DOI:10.1021/acs.joc.0c00022 |
| [276] |
A. Hirose, A. Watanabe, K. Ogino, et al., J. Am. Chem. Soc. 143 (2021) 12387-12396. DOI:10.1021/jacs.1c06450 |
| [277] |
M. Fadel, E.M. Carreira, J. Am. Chem. Soc. 145 (2023) 8332-8337. DOI:10.1021/jacs.3c02113 |
| [278] |
G.H. Stout, W.J. Balkenhol, M. Poling, et al., J. Am. Chem. Soc. 92 (1970) 1070-1071. DOI:10.1021/ja00707a058 |
| [279] |
A. Ronlán, B. Wickberg, Tetrahedron Lett. 49 (1970) 4261-4264. |
| [280] |
S.M. Kupchan, R.L. Baxter, Science 187 (1975) 652-653. DOI:10.1126/science.1114315 |
| [281] |
K. Sakata, K. Kawazu, T. Mitsui, Agr. Biol. Chem. 35 (1971) 1084-1091. |
| [282] |
K. Sakata, K. Kawazu, T. Mitsui, Agr. Biol. Chem. 35 (1971) 2113-2126. |
| [283] |
M. Hergenhahn, W. Adolf, E. Hecker, Tetrahedron Lett. 16 (1975) 1595-1598. |
| [284] |
R.G. Powell, D. Weisleder, C.R. Smith Jr, J. Nat. Prod. 48 (1985) 102-107. DOI:10.1021/np50037a018 |
| [285] |
W. Adolf, B. Sorg, M. Hergenhahn, et al., J. Nat. Prod. 45 (1982) 347-354. DOI:10.1021/np50021a018 |
| [286] |
S.M. Kupchan, Y. Shizuri, T. Murae, et al., J. Am. Chem. Soc. 98 (1976) 5719-5720. DOI:10.1021/ja00434a063 |
| [287] |
S. Zayed, W. Adolf, A. Hafez, E. Hecker, Tetrahedron Lett. 18 (1977) 3481-3482. DOI:10.1016/S0040-4039(01)83271-8 |
| [288] |
W. Adolf, E.H. Seip, E. Hecker, S.F. Dossaji, J. Nat. Prod. 51 (1988) 662-674. DOI:10.1021/np50058a003 |
| [289] |
W. He, M. Cik, L. Van Puyvelde, et al., Bioorg. Med. Chem. 10 (2002) 3245-3255. DOI:10.1016/S0968-0896(02)00163-3 |
| [290] |
Y. Asada, K. Otsuki, M. Morooka, et al., J. Nat. Prod. 85 (2022) 2399-2405. DOI:10.1021/acs.jnatprod.2c00618 |
| [291] |
K. Otsuki, T. Kobayashi, K. Nakamura, et al., Fitoterapia 172 (2024) 105731. DOI:10.1016/j.fitote.2023.105731 |
| [292] |
H. Jayasuriya, D.L. Zink, S.B. Singh, et al., J. Am. Chem. Soc. 122 (2000) 5719-5720. |
| [293] |
S.G. Liao, H.D. Chen, J.M. Yue, Chem. Rev. 109 (2009) 1092-1140. DOI:10.1021/cr0782832 |
| [294] |
A. Szallasi, P.M. Blumberg, Life Sci. 47 (1990) 1399-1408. DOI:10.1016/0024-3205(90)90518-V |
| [295] |
A. Szallasi, P.M. Blumberg, Pharmacol. Rev. 51 (1999) 159-212. |
| [296] |
I. Kissin, A. Szallasi, Curr. Top Med. Chem. 11 (2011) 2159-2170. DOI:10.2174/156802611796904924 |
| [297] |
M. Aghazadeh Tabrizi, P.G. Baraldi, S. Baraldi, et al., Med. Res. Rev. 37 (2017) 936-983. DOI:10.1002/med.21427 |
| [298] |
J.I. Kang, J.Y. Hong, H.J. Lee, et al., PLoS One 10 (2015) e0144368. DOI:10.1371/journal.pone.0144368 |
| [299] |
M.O. Villareal, Y. Sato, K. Matsuyama, H. Isoda, BMC Cancer 18 (2018) 902. DOI:10.1186/s12885-018-4805-8 |
| [300] |
J. Wu, L. Guo, X. Qiu, et al., Br. J. Cancer 123 (2020) 1673-1685. DOI:10.1038/s41416-020-01085-z |
| [301] |
C.S. Fermaintt, T. Peramuna, S. Cai, et al., Cancers 13 (2021) 2834. DOI:10.3390/cancers13112834 |
| [302] |
J.L. Huang, X.L. Yan, W. Li, et al., J. Am. Chem. Soc. 144 (2022) 17522-17532. DOI:10.1021/jacs.2c06449 |
| [303] |
G. Moshiashvili, N. Tabatadze, V. Mshvildadze, Fitoterapia 143 (2020) 104540. DOI:10.1016/j.fitote.2020.104540 |
| [304] |
Y.W. Nie, Y. Li, L. Luo, et al., Molecules 26 (2021) 6598. DOI:10.3390/molecules26216598 |
| [305] |
Z.L. Hou, G.D. Yao, S.J. Song, Chin. Herb. Med. 13 (2021) 145-156. |
| [306] |
C. Bailly, Biomolecules 12 (2022) 192. DOI:10.3390/biom12020192 |
| [307] |
V. Vidal, O. Potterat, S. Louvel, et al., J. Nat. Prod. 75 (2012) 414-419. DOI:10.1021/np200855d |
| [308] |
L. Huang, P. Ho, J. Yu, et al., PLoS One 6 (2011) e26677. DOI:10.1371/journal.pone.0026677 |
| [309] |
W. Lai, L. Huang, L. Zhu, et al., J. Med. Chem. 58 (2015) 8638-8646. DOI:10.1021/acs.jmedchem.5b01233 |
| [310] |
Y. Gao, H. Fan, A. Nie, et al., J. Ethnopharmacol. 293 (2022) 115270. DOI:10.1016/j.jep.2022.115270 |
| [311] |
Y. Shen, W.J. Liang, Y.N. Shi, et al., Nat. Prod. Rep. 37 (2020) 763-796. DOI:10.1039/d0np00002g |
| [312] |
L.J. Chen, X.C. Tang, M.Y. Wang, et al., Chin. J. Pharmacol. Toxicol. 1 (1987) 123-128. |
| [313] |
H. Xu, S. Yao, Drugs Clin. 38 (2023) 2639-2644. |
| [314] |
M. Ono, T. Satoh, Jpn, J. Pharmacol. 55 (1991) 523-530. DOI:10.1254/jjp.55.523 |
| [315] |
X. Huang, Y. Yang, J. Zhu, et al., Basic Clin. Pharmacol. Toxicol. 104 (2009) 145-154. DOI:10.1111/j.1742-7843.2008.00357.x |
| [316] |
X.X. Liu, X.X. Jian, X.F. Cai, et al., Chem. Pharm. Bull. 60 (2012) 144-149. DOI:10.1248/cpb.60.144 |
| [317] |
F.P. Wang, Q.H. Chen, X.Y. Liu, Nat. Prod. Rep. 27 (2010) 529-570. DOI:10.1039/b916679c |
| [318] |
Y. Wu, S. Shao, Q. Guo, et al., Org. Lett. 21 (2019) 6850-6854. DOI:10.1021/acs.orglett.9b02479 |
| [319] |
S. Shao, H. Xia, M. Hu, et al., J. Neuroinflammation 17 (2020) 13. DOI:10.1186/s12974-019-1696-9 |
| [320] |
F. Wang, Z. Yue, P. Xie, et al., Molecules 21 (2016) 1175. DOI:10.3390/molecules21091175 |
| [321] |
J. Yu, T.P. Yin, J.P. Wang, et al., Nat. Prod. Res. 31 (2016) 228-232. |
| [322] |
B. Song, B. Jin, Y. Li, et al., Molecules 23 (2018) 1108. DOI:10.3390/molecules23051108 |
| [323] |
H. Ahmad, S. Ahmad, S.A.A. Shah, et al., Bioorg. Med. Chem. 25 (2017) 3368-3376. DOI:10.1016/j.bmc.2017.04.022 |
| [324] |
J. Wang, X.H. Meng, T. Chai, et al., Nat. Prod. Bioprospect. 9 (2019) 419-423. DOI:10.1007/s13659-019-00227-y |
| [325] |
Y. Li, J. Zeng, Y.H. Tian, et al., Phytochemistry 190 (2021) 112880. DOI:10.1016/j.phytochem.2021.112880 |
| [326] |
L.H. Shan, L. Chen, F. Gao, et al., Nat. Prod. Res. 33 (2018) 3254-3259. |
| [327] |
S. Huang, Y. Lv, J.Z. Wang, et al., Planta Med. 89 (2022) 674-682. |
| [328] |
L. Mi, Y.C. Li, M.R. Sun, et al., Chin. J. Nat. Med. 19 (2021) 505-520. |
| [329] |
Y.Z. Li, Y.S. Shang, X.H. Li, et al., Eur. J. Med. Chem. 243 (2022) 114776. DOI:10.1016/j.ejmech.2022.114776 |
| [330] |
L.X. Wan, J.F. Zhang, Y.Q. Zhen, et al., J. Nat. Prod. Res. 84 (2021) 1067-1077. DOI:10.1021/acs.jnatprod.0c01111 |
| [331] |
Y. Zhang, T.J. Zhang, X.Y. Li, et al., Eur. J. Med. Chem. 210 (2021) 114776. |