 2024, Vol. 35
2024, Vol. 35 


Phosphoinositide 3-kinases (PI3Ks) are a kind of intracellular phosphatidylinositol kinases with serine/threonine (Ser/Thr) kinase activity. PI3Ks are divided into three classes, of which class Ⅰ is the most widely studied. Class I PI3Ks are heterodimers composed of a regulatory subunit and a catalytic subunit. The regulatory subunits contain the SH2 domain that is responsible for binding to the catalytic subunits, among which p85α and p85β are broadly expressed. Moreover, there are four types of catalytic subunits, namely p110α, p110β, p110γ and p110δ, which regulate downstream signal transduction with the coordination of regulatory subunits [1]. Therefore, PI3Ks play a prominent role in the processes of cell proliferation, differentiation, apoptosis, cytoskeleton construction, and glucose metabolism through participating in the PI3K-protein kinase B (AKT)-mammalian target of rapamycin (mTOR) signal pathway. Hyperactivation of this pathway caused by PI3Ks mutation or overexpression is a universal reason for tumorigenesis, especially for the breast cancer [2]. Despite the enormous demand and prospect of drug discovery targeting PI3Ks, there are few drugs approved for clinical treatment [3]. In general, the major challenges in function identification and drug development of PI3K are as follows: (1) Currently, inhibitors of PI3K act on the relatively conserved kinase domain of p110 catalytic subunits, causing the poor selectivity of drugs that is responsible for the toxicity and limitation in clinical. (2) In addition to regulating the enzymatic activity of p110s, p85 regulatory subunits possess other non-enzymatic functions [4-8]. To investigate the whole functions of PI3K, simultaneously blocking the activity of p110s and p85s is required [9]. While no effective modulators of p85s have been developed due to the lack of binding pockets, furthermore, the technology of gene editing sometimes suffers from unexpectedly compensatory effects. Therefore, it is particularly necessary to develop the selective small-molecule tools for PI3K that can regulate the functions of p110s and p85s concurrently.
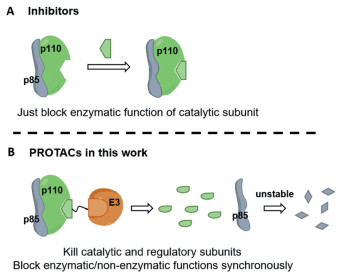
Proteolysis-targeting chimeras (PROTAC) is a novel technology to induce the degradation of target protein on the strength of natural ubiquitin-proteasome system, by using a bifunctional small molecule to recruit the specific E3 ligase to ubiquitinate the target proteins [10-14]. PROTAC has been broadly applied to achieve the degradation of diverse targets, PI3K is also included [15-17]. However, the reported degraders targeting PI3K only possess low efficiency of degradation which are not appropriate chemical probes for related research [18,19]. So far, no PROTAC molecules that can eliminate the functions of p85 regulatory subunits have been reported. To our knowledge, a selective and potent PI3K-α degrader targeting PI3K-α has not yet been reported. Therefore, we developed a highly efficient PROTAC molecule that selectively degraded p110α and abrogated the function of p85 regulatory subunits (Fig. 1). This work puts forward the first small molecule degraders which can modulate both kinase and non-kinase functions of PI3K-α.

|
Download:
|
| Fig. 1. Schema of PROTAC molecules and inhibitors targeting PI3K. (A) Existing inhibitors targeting PI3Ks could only block the catalytic function of the kinase. (B) In this work, we synthesized the selective degrader targeting PI3K-α which degraded the catalytic and regulatory subunits simultaneously. | |
{kind=link}
To develop degraders of PI3K-α, the PI3K-targeting arm was naturally conjugated to the E3 ligase ligand through different types of linkers. Diverse PI3K ligands and E3 ligases, including cereblon (CRBN) ligands and Von Hippel-Lindau (VHL), were tried based on our designing principles [20,21]. Through screening in the breast cancer cell line MDA-MB-231, we found that the degradation of p110α induced by CRBN is more efficient than VHL (Figs. S1C and D in Supporting information). Perhaps due to that CRBN tends more to form the stable ternary complex with PI3K proteins and PROTAC molecules under the specific linkers and proteins of interest (POI) binders that we used. Among the hit compounds, an Food and Drug Administration (FDA)-approved pan-inhibitor copanlisib proved to be a suitable POI binder for achieving notable degradation of targeted protein. As shown in Fig. 2A, based on the principle that the linkage site is supposed to extend to solvent zone and possesses no binding force with target protein (pointed out with red arrow in Fig. 2C), the terminal morpholine group of copanlisib was connected with the CRBN binder pomalidomide through different lengths of linkers, generating the molecules ZM-PI01, ZM-PI02, ZM-PI03 and ZM-PI04.

|
Download:
|
| Fig. 2. Design and screening of degraders. (A, B) Structure and degradation efficiency of the PROTAC molecules. The column diagram showed the statistical results of immunoblots in MDA-MB-231 cells after treatment with the degraders for 24 h. (C) Binding model of ZM-PI05 (yellow sphere) with DDB1-CRBN and p110α/p85α (referenced PDB files: 4CI3, 5G2N, 5ITD). The red arrow represents linkage site of copanlisib that extends to the solvent zone. The black arrows represent the narrow gate of p110α that cannot accommodate large groups, such as triazole. | |
{kind=link}
To our delight, the first-generation molecules could efficiently degrade the p110α isoform and slightly down-regulate p110γ protein, without affecting the protein level of p110β which is highly homologous to p110α. ZM-PI02 was found to be the most potent degrader (Fig. 2A). It is known that p110α isoform plays an important role in breast cancers [22]. Therefore, we attempted to optimize the hit molecule, looking forward to obtaining the degraders with better selectivity and efficiency on p110α. Through analyzing the structure of p110α protein complexed with copanlisib, we find that the gate region of ATP-binding pocket in p110α protein is relatively narrow, which exactly accommodates the large morpholine ring of copanlisib. In the design of first-generation PROTAC molecules, morpholine group was chosen as the linkage site. We speculated that connection with an extra linker maybe restrict the freedom of morpholine ring in conformation, generating a large clash between the degrader and the gate region of p110α, which is extremely unfavourable for the binding of PROTAC molecules to p110α proteins. Previous SAR studies have shown that the morpholine ring at the end of copanlisib makes no contribution to the binding affinity with proteins, which is only supposed to modulate the water solubility and metabolic stability of the inhibitor. Based on the above speculations, we decided to cut off the morpholine tail to improve the efficiency of degraders. It deserves to be mentioned that the degraders with a triethylene glycol linker could induce the degradation p110α more efficiently, according to the evaluation results of the first-generation degraders. Hence, the similar lengths of linkers were introduced to the core structure of copanlisib, producing the second-generation degraders (Fig. 2B).
After evaluation on MDA-MB-231 cell line, the second-generation molecules were identified as degraders targeting p110α protein with the increased efficiency by two fold (degradation ratio (DR) at 10 nmol/L of ZM-PI02 = 25%, DR at 10 nmol/L of ZM-PI05 = 65%), while possessing little effect on the protein level of p110β and p110γ (Fig. 2B). Obviously, the strategy of abrogating morpholine group is resultful for enhancing the degradation efficiency and retaining selectivity of the degraders, which may be related to the increased rotational freedom of the molecules. To further validate the conjecture, we changed the position of triazole in the linkers to obtain the molecules ZM-PI09/ZM-PI10 and ZM-PI11/ZM-PI12. By comparison, the closer the triazole was to the target protein, the less efficient the degraders were (half-maximal degradation concentration (DC50) of ZM-PI09/ZM-PI10 was about 50 nmol/L; DC50 of ZM-PI11/ZM-PI12 was about 20 nmol/L, Fig. S1B in Supporting information), indicating that to degrade p110α protein, the certain freedom was required for the part of linker near to the targeting pocket, because of the narrow gate of p110α protein which is hard to accommodate a large group in the ternary complex (pointed out with black arrows in Fig. 2C). While the suitable angle of the specific linker that caused by triazole maybe another factor.
Copanlisib is a pan-inhibitor which displays high binding affinity on class Ⅰ p110s, while we did not observe strong down-regulation of p110β and p110γ proteins after treatment with the representative degrader ZM-PI05. The reason may be that ZM-PI05 could recruit CRBN to get close to p110α protein rather than p110β or p110γ and induce the formation of ternary complex. In fact, it displayed a definite advantage of PROTAC in selectivity. Among the second-generation degraders, ZM-PI05 induced obvious degradation of p110α and showed much stronger anti-proliferative activity, which was used in the subsequent studies.
Firstly, the degradation efficiency of ZM-PI05 against p110α protein was further tested on MDA-MB-231. It showed that ZM-PI05 degraded p110α in a dose-dependent manner, with a DC50 value of 5 nmol/L (Fig. 3A). Although the protein level of p110β was slightly down-regulated at high concentrations, ZM-PI05 was still more potent to induce the degradation of p110α, showing that the degrader in this work possesses quite good selectivity. The degradation of p110α was quite fast, taking only 2 h after treatment with the degrader at 100 nmol/L (Fig. 3B). Besides p110α, we also examined the protein level of regulatory subunit p85s. Encouragingly, we found when treated with the degrader for more than 12 h, down-regulation of the regulatory subunit p85 proteins could be observed (Fig. 3B). In comparison, the downregulation of p85 proteins cannot be induced by PI3K inhibitors. As we know, several PROTAC molecules targeting p110α has been reported, so we synthesized the degrader named compound D according to the literature [18] and tested its efficiency. Compound D almost cannot induce the degradation of p85 proteins even at the concentration of 10 μmol/L, while ZM-PI05 induced obvious degradation of p85 proteins at the lower concentrations, showing that ZM-PI05 is a more potent degrader of PI3K-α (Fig. S3B in Supporting information). It was reported that in the natural state, the catalytic subunit p110s form various complexes with the regulatory subunit p85s to regulate the enzymatic activity and stability of p110 and p85 proteins [23,24]. When we knocked down p110α proteins with short hairpin RNA (shRNA), p85α and p85β will also be down-regulated subsequently (Fig. S4A in Supporting information), which is consistent with previous findings [25]. Also, the CHX chase experiment indicated that the half-life of p85 proteins in MDA-MB-231 cells was significantly decreased if the p110α protein was knocked down (Figs. S4B and C in Supporting information).Therefore, we speculated that the downregulation of p85 protein was the secondary effect of p110α degradation, while not caused by ZM-PI05 directly. Generally, PI3K inhibitors could only block the enzymatic function of catalytic subunit p110s. While the PROTAC molecules that we developed regulated the enzymatic and non-enzymatic functions of p110α and p85 by degrading them simultaneously, providing a powerful tool compound for the non-enzymatic functional study of PI3K-α.

|
Download:
|
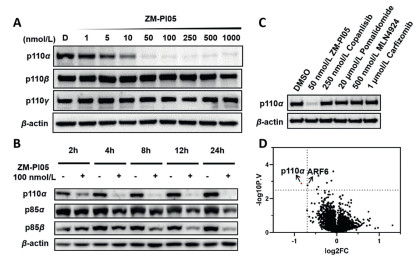
| Fig. 3. Degradation characterization of ZM-PI05. (A) Immunoblots for p110α, p110β and p110γ proteins in MDA-MB-231 cells after treatment with ZM-PI05 for 6 h. (B) Immunoblots for p110α, p85α and p85β in MDA-MB-231 cells treated with 100 nmol/L ZM-PI05 at indicated time points. (C) Immunoblot analysis of p110α in MDA-MB-231 cells pre-treated with DMSO, copanlisib (250 nmol/L), pomalidomide (20 μmol/L) or MLN-4924 (500 nmol/L), carfilzomib (1 μmol/L) for 4 h, and then treated with ZM-PI05 (50 nmol/L) for 12 h. (D) Proteomic analysis of proteins in MDA-MB-231 cells treated with 100 nmol/L ZM-PI05 for 5 h (n = 3). Volcano plots of −log10 (P value) versus log2 Fold Change. P values were calculated using two tailed unpaired Student's t-test. | |
{kind=link}
In addition, the effect of ZM-PI05 on the whole-proteome was analyzed, we were delighted to find that ZM-PI05 displayed excellent selectivity on p110α, with no significant degradation of other isoforms (Fig. 3D). Unexpectedly, ARF6 was found to be down-regulated in proteomic analysis, which was confirmed as a false positive result by Western blot assay under the same treatment conditions (Fig. S3C in Supporting information). After determining the degradation efficiency of ZM-PI05 on p110α, rescue assays were carried out to verify whether the degrader acted as a PROTAC molecule. When pretreated with the inhibitor copanlisib or pomalidomide to block the pockets of p110α or CRBN, ZM-PI05 could not induce the degradation of p110α as usual (Fig. 3C), which showed that the ligands at both ends of the degrader were necessary to recruit the target protein and E3 ligase simultaneously. Furthermore, synchronous treatment with the proteasome inhibitor carfilzomib caused the same result (Fig. 3C), indicating that the degradation of p110α induced by ZM-PI05 occurred based on ubiquitin-proteasome system.
To sum up, we obtained a highly efficient and p110α-selective degrader ZM-PI05 through two rounds of design and optimization, the degradation mechanism study showed that the molecule exactly acted as a PROTAC to induce the degradation of p110 protein by UPS, and subsequently down-regulated the p85 regulatory subunits.
Next, the effect of ZM-PI05 was evaluated in various triple-negative breast cancer cells MDA-MB-231, MDA-MB-468 and non-triple-negative breast cancer cells MDA-MB-453, SK-BR-3, BT474, MCF7. The results showed that ZM-PI05 could stably degrade p110α protein in these cell lines (Fig. 4D). Accordingly, the degrader has much stronger inhibitory effect on growth of these tumor cells than non-selective inhibitor copanlisib, half-maximal inhibitory concentration (IC50) was all below 100 nmol/L (Fig. 4A).

|
Download:
|
| Fig. 4. Inhibitory effect of ZM-PI05 in breast cancer cells. (A) Half inhibition concentrations of copanlisib and ZM-PI05 in multiple breast cancer cells. (B) Anti-proliferative activity on MDA-MB-231 and SK-BR-3 cells treated with copanlisib or ZM-PI05 for 72 h (n = 3). (C) Immunoblots for p110, p85 and actin proteins in MDA-MB-231 cells after treatment with ZM-PI05 for 24 h. (D) Immunoblots for p110α in MDA-MB-468, MDA-MB-453, MCF7, SK-BR-3 and BT474 cells after treatment with ZM-PI05 for 24 h (n = 2). The data are presented as the mean ± standard deviation (SD) values. The statistical values of p110α protein were calculated with GraphPad Prism8. | |
{kind=link}
Phosphorylation of AKT is the most important down-stream signal of PI3K, so the level of p-AKT was detected to verify the effect of degraders on PI3K-AKT pathway. ZM-PI05 strongly inhibited the phosphorylation of AKT proteins, indicating that the potency of ZM-PI05 is corresponding to the enzymatic function of PI3K (Fig. S3D in Supporting information).
Furthermore, the inhibitory mechanism of ZM-PI05 on breast cancer cells was explored. It was observed that for non-triple-negative breast cancer cells, ZM-PI05 significantly induced apoptosis at the concentration of 250 nmol/L, which was stronger than copanlisib (Figs. S5C and S6 in Supporting information). Accordingly, the degrader down-regulated PI3K-related anti-apoptotic proteins XIAP and MCL-1 (Fig. S5D in Supporting information). However, apoptosis did not occur in triple-negative breast cancer cells in presence of the degrader ZM-PI05, while cell cycle arrest in G1 phase was observed (Figs. S5B and S7A, C, D in Supporting information), which was further confirmed by the downregulation of cyclinD (Fig. S7B in Supporting information).
The results above proved that ZM-PI05 displayed different phenotypes in multiple breast cancer cells. In non-triple-negative breast cancer cells, ZM-PI05 induced significant cell apoptosis, while the degrader inhibited growth of triple-negative breast cancer cells mainly by arresting the cell cycle.
It showed that the degrader ZM-PI05 induced about 50% decrease of p85 expression in different breast cancer cells (Fig. S9A in Supporting information). p85α and p85β are two types of regulatory subunits related to p110α, which paly different roles in the proliferation of tumor cells [26]. So, in order to investigate which regulatory subunit was related to the anti-proliferation activity of ZM-PI05, we utilized shRNA to construct p85α and p85β knockdown MDA-MB-231 cells, respectively. The results showed that when p85β was knocked down, the growth of tumor cells was significantly slowed down, while p85α knockdown had little effect on cell proliferation (Fig. S9B in Supporting information). Furthermore, we found that deletion of p85β resulted in decreased sensitivity of tumor cells to the degrader. Compared with MDA-MB-231 cells which were transfected with blank plasmid, p85β knockdown cells were less sensitive to ZM-PI05, while p85α knockdown cells still had response to the molecule. The above results indicated that down-regulation of p85β rather than p85α was the main reason why the degrader displayed such better proliferation inhibitory activity than the inhibitor, which was also consistent with the reported research results [27].
Additionally, we further investigated the p85β-related functions affected by ZM-PI05. The iSH2, cSH2, and nSH2 domains at the N-terminus of p85 protein bind to p110 proteins to regulate its activity, while SH3 and BH domains at the C-terminus are important for its regulatory functions independent of PI3K. For example, XBP-1 is translocated to the nucleus with the assistant of p85 proteins to regulate the expression of various downstream proteins, and then unfolded protein response (UPR) occurs [28,29]. The PROTAC molecule ZM-PI05 induced the degradation of p85β protein, inevitably blocking the non-enzymatic functions of its backbone, such as transcription regulation. To explore whether ZM-PI05 could affect the above functions by degrading p85β protein, the change of transcription process was detected by RNA-seq.
Transcriptomics revealed that the ZM-PI05 group showed a distinct transcriptional profile compared to DMSO, copanlisib or p85β-knockdown groups (Figs. S8D and E in Supporting information). Compared with copanlisib group, ZM-PI05 caused significantly deeper and broader transcriptional expression changes, with 801 genes to be up-regulated and 587 genes to be down-regulated (Fig. S9C in Supporting information). While ZM-PI05 and p85β knockdown groups showed the common transcriptional disruption on 160 genes (Fig. S9D in Supporting information), which were enriched in angiogenesis and stress response and cellular adhesion pathways (Fig. S8B in Supporting information). This finding is consistent with the existing studies that p85β has multiple independent functions involved in cytoskeleton and cell adhesion [30]. Also, the azide intermediate 17a composed of a CRBN binder and linker that are used in the synthesis of ZM-PI05 showed slight effect on gene expression (Fig. S8C in Supporting information). These results proved that ZM-PI05 affected the non-enzymatic functions of the PI3Kα complex, while the binders at both end did not. Investigation will be conducted to reveal more biological details in our future study.
In conclusion, we developed the PROTAC molecule ZM-PI05 that can selectively degrade p110α and down-regulate p85 for the first time. In breast cancer cell lines, ZM-PI05 efficiently degraded p110α proteins, accordingly displayed stronger inhibition effect than the inhibitor on these cells. ZM-PI05 affected the proliferation of non-triple-negative breast cancer cells and triple-negative breast cancer cells by inducing apoptosis and cell cycle arresting, respectively. In addition, the degrader ZM-PI05 down-regulated p85β protein concurrently, which is the regulatory subunit of p110s to affect the overall functions of PI3K and enhance the anti-proliferation efficiency of the degrader. Because of this, PROTAC molecules can achieve equivalent or even better inhibitory effects than inhibitors at the lower concentrations, that is, selective PI3K degraders can be used in clinical treatment at low doses to avoid the reported toxicity caused by current drugs. The overexpression of p85β frequently occurs in breast cancer cells, so it is also a target worthy to be studies. Currently, there is no modulators targeting p85β, and the existing degraders targeting p110α cannot down-regulate p85 proteins efficiently. The PROTAC molecule ZM-PI05 developed in this work could simultaneously down-regulate the subunits p85α and p85β, which provided a novel and useful tool compound for the functional studies of p85 regulatory subunits in further in-depth biomedical research.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by National Key R&D Program of China (Nos. 2021YFA1302100, 2021YFA1300200, 2020YFE0202200), National Natural Science Foundation of China (Nos. 82125034, 82330115).
The authors acknowledge Profs. Wei Wu, Jianyang Zeng, Haiteng Deng and Haitao Li from Tsinghua University for academic suggestions and Dr. Xiuyun Sun from Tsinghua University for her help.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109577.
| [1] |
M.D. Goncalves, B.D. Hopkins, L.C. Cantley, N. Engl. J. Med. 379 (2018) 2052-2062. DOI:10.1056/nejmra1704560 |
| [2] |
M.P. Lux, P.A. Fasching, M.G. Schrauder, et al., Breast Care 11 (2016) 398-404. DOI:10.1159/000453133 |
| [3] |
X.B. Zhang, X.R. Li, J. Zhang, Curr. Cancer Drug Targets 13 (2013) 175-187. DOI:10.2174/1568009611313020007 |
| [4] |
M.D. Chamberlain, T.R. Berry, M.C. Pastor, D.H. Anderson, J. Biol. Chem. 279 (2004) 48607-48614. DOI:10.1074/jbc.M409769200 |
| [5] |
Z. Garcia, V. Silio, M. Marques, et al., EMBO J. 25 (2006) 4740-4751. DOI:10.1038/sj.emboj.7601324 |
| [6] |
M.E. Urick, M.L. Rudd, A.K. Godwin, et al., Cancer Res. 71 (2011) 4061-4067. |
| [7] |
L.W.T. Cheung, S.X. Yu, D. Zhang, et al., Cancer Cell 26 (2014) 479-494. DOI:10.1016/j.ccell.2014.08.017 |
| [8] |
A.E. Cariaga-Martínez, I. Cortés, E. García, et al., Biol. Open. 3 (2014) 924-936. DOI:10.1242/bio.20148185 |
| [9] |
A.B. Hanker, V. Kaklamani, C.L. Arteaga, Cancer Discov. 9 (2019) 482-491. DOI:10.1158/2159-8290.cd-18-1175 |
| [10] |
K.M. Sakamoto, K.B. Kim, R. Verma, et al., Mol. Cell. Proteomics 2 (2013) 1350-1358. |
| [11] |
M. Pettersson, C.M. Crews, Drug Discov. Today Technol. 31 (2019) 15-27. DOI:10.1016/j.ddtec.2019.01.002 |
| [12] |
A. Hanzl, G.E. Winter, Curr. Opin. Chem. Biol. 56 (2020) 35-41. DOI:10.1016/j.cbpa.2019.11.012 |
| [13] |
A. Chen, Y. Zhong, Y. Liu, et al., Chin. Chem. Lett. 34 (2023) 107923. DOI:10.1016/j.cclet.2022.107923 |
| [14] |
Y.H. Sun, X. Luo, Z.M. Yang, et al., Chin. Chem. Lett. 34 (2023) 107924. DOI:10.1016/j.cclet.2022.107924 |
| [15] |
G.M. Burslem, C.M. Crews, Cell 181 (2020) 102-114. DOI:10.1016/j.cell.2019.11.031 |
| [16] |
M. He, C.G. Cao, Z.H. Ni, et al., Signal. Transduct. Target. Ther. 7 (2022) 181. DOI:10.1038/s41392-022-00999-9 |
| [17] |
B. Dale, M. Cheng, K. Park, et al., Nat. Rev. Cancer 21 (2021) 638-654. DOI:10.1038/s41568-021-00365-x |
| [18] |
W.L. Li, C.M. Gao, L. Zhao, et al., Eur. J. Med. Chem. 151 (2018) 237-247. DOI:10.1016/j.ejmech.2018.03.066 |
| [19] |
H.L. Wang, C.C. Li, X.Q. Liu, M.L. Ma, Bioorg. Med. Chem. 61 (2022) 116707. DOI:10.1016/j.bmc.2022.116707 |
| [20] |
S. Su, Z.M. Yang, H.Y. Gao, et al., J. Med. Chem. 62 (2019) 7575-7582. DOI:10.1021/acs.jmedchem.9b00871 |
| [21] |
L.G. Wang, X.J. Shao, T.B. Zhong, et al., Nat. Chem. Biol. 17 (2021) 567-575. DOI:10.1038/s41589-021-00742-5 |
| [22] |
J.J. Zhao, H. Cheng, S. Jia, et al., Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 16296-16300. DOI:10.1073/pnas.0607899103 |
| [23] |
B. Geering, P.R. Cutillas, B. Vanhaesebroeck, Biochem. Soc. Trans. 35 (2007) 199. DOI:10.1042/BST0350199 |
| [24] |
M. Fox, H.R. Mott, D. Owen, Biochem. Soc. Trans. 48 (2020) 1397-1417. DOI:10.1042/bst20190845 |
| [25] |
S.M. Brachmann, K. Ueki, J.A. Engelman, R.C. Kahn, L.C. Cantley, Mol. Cell. Biol. 25 (2005) 1596-1607. DOI:10.1128/MCB.25.5.1596-1607.2005 |
| [26] |
J. Vallejo-Díaz, M. Chagoyen, M. Olazabal-Morán, A. González-García, A.C. Carrera, Trends Cancer 5 (2019) 233-244. DOI:10.1016/j.trecan.2019.02.009 |
| [27] |
I. Cortes, J. Sanchez-Ruiz, S. Zuluaga, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 11318-11323. DOI:10.1073/pnas.1118138109 |
| [28] |
S.W. Park, Y. Zhou, J. Lee, et al., Nat. Med. 16 (2010) 429-437. DOI:10.1038/nm.2099 |
| [29] |
J.N. Winnay, J. Boucher, M.A. Mori, K. Ueki, C.R. Kahn, Nat. Med. 16 (2010) 438-445. DOI:10.1038/nm.2121 |
| [30] |
L. Rao, V.C.Y. Mak, Y. Zhou, et al., Nat. Commun. 11 (2020) 2291. DOI:10.1038/s41467-020-16061-7 |