 2024, Vol. 35
2024, Vol. 35 


b Jiangsu Province Geriatric Hospital, Nanjing 210024, China;
c School of Allied Medical Sciences, Delhi Skill and Entrepreneurship University, and Department of Biomedical Science, Acharya Narendra Dev College, University of Delhi, New Delhi 110019, India;
d Department of Chemistry, University of Massachusetts Boston, Boston, MA 02125, United States;
e V.P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry, The National Academy of Sciences of Ukraine, Kyiv 02094, Ukraine;
f Department of Chemistry, University of Hamburg, Martin-Luther-King-Platz 6, Hamburg 20146, Germany;
g Department of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizábal 3, San Sebastián 20018, Spain;
h IKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, Plaza Bizkaia, Bilbao 48013, Spain
Since the approval of fludrocortisone [1,2], the first fluorine-containing pharmaceutical, synthesis, and biological evaluation of fluorinated compounds has been one of the most fast-growing field of multidisciplinary research [3–20]. It is rather remarkable that fluorine-containing compounds now constitute around a quarter of small molecule drugs in the pharmaceutical market [21–38]. Furthermore, fluorinated derivatives are usually found among the most prescribed and top-selling drugs [39,40]. Fluorine-editing and fluorine-scans are now routine stages in drug development, having a significant influence on organic synthesis and medicinal chemistry. Practitioners in these fields are increasingly turning their focus towards innovative approaches to incorporating fluorine and fluorine-containing motifs into organic chemistry scaffolds.
In the past five years, more than fifty fluorine-containing small molecule drugs were approved by FDA for the treatment of several kinds of diseases. In 2018, seventeen fluorine-containing drugs were approved, which feature twelve aromatic fluorine, six aromatic CF3, and one aliphatic CF2 substitutions [27]. These drugs were used for the treatment of chronic immune thrombocytopenia, melanoma, intra-abdominal infections, lung cancer, breast cancer, influenza, smallpox infections, chemotherapy-induced nausea and vomiting, (human immunodeficiency virus) HIV, malarial, pain associated with endometriosis, prostate cancer, cystic fibrosis and myeloid leukemia. In 2019, eleven fluorine-containing drugs were approved [28], which feature five aromatic fluorine, three aromatic CF3, three aliphatic CF3, and one aromatic CF3O substituents. These drugs were approved for the therapeutic areas, such as schizophrenia, migraine, multiple sclerosis, insomnia, rheumatoid arthritis, anti-tuberculosis, breast cancer, lymphoma kinase inhibitor, and serotonin receptor antagonist. In 2020, thirteen fluorine-containing small molecule drugs were approved [41], and the fluorine-containing substituents include eleven aromatic fluorine, one aromatic CF3, one aliphatic CHF, and one CF2 groups. These drugs were developed for the treatment of Cushing’s disease, neurofibromatosis, myelodysplastic syndromes, lung cancer, advanced gastrointestinal stromal tumor, migraine, Alzheimer’s disease, myelodysplastic syndromes, and hereditary angioedema attacks. In 2021, nine new fluorine-containing drugs were approved. These small molecular drugs feature eight aromatic fluorine, one aliphatic CHF, one aromatic CF3, one aliphatic CF3, and one CClF2 groups. The therapeutic areas of these fluorine-containing drugs include multiple myeloma, lymphoma, HIV, chronic heart failure, chronic myeloid leukemia, anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis, migraines, von Hippel-Lindau disease, and non-small cell lung cancer [42]. In 2022, four new fluorine-containing drugs were approved [35], which feature three aromatic fluorine, two aliphatic CF2, one alkenyl CF groups, one aromatic CF3, and two aliphatic CF3 substituents. The therapeutic areas included non-small cell lung cancer, HIV, recurrent vulvovaginal candidiasis, helicobacter pylori (Hp) infection. As presented above, there are some interesting trends in the drugs containing fluoro/fluoroalkyl groups. Aromatic fluorine is most used moiety in these drugs, and a high percentage of the related drugs were observed each year. Also, several kinds of aliphatic fluoro groups have been introduced into the drugs in very recent years, including CHF, CF2, CClF2, and so on. On the other hand, an increased proportion of chiral fluorinated drugs were approved in enantiomerically pure form, and even the chiral C-F center (belzutifan) was observed in 2021.
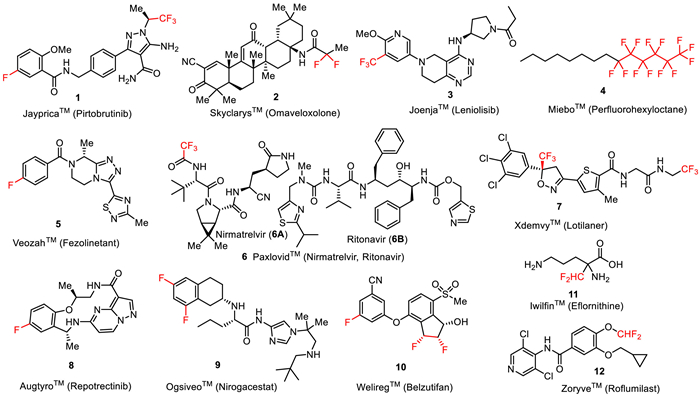
To help synthetic organic chemists to navigate their choice of targets and methodological approaches, we continue to provide timely updates on the newly approved drugs appearing on the pharmaceutical market [41,42]. The goal of this review article is to profile twelve fluorine-containing drugs 1–12 introduced to the market by the FDA during the current year (Fig. 1). In particular, we will present JaypricaTM (Pirtobrutinib) (1), SkyclarysTM (Omaveloxolone) (2), JoenjaTM (Leniolisib) (3), MieboTM (Perfluorohexyloctane) (4), VeozahTM (Fezolinetant) (5), PaxlovidTM (Nirmatrelvir and Ritonavir) (6), XdemvyTM (Lotilaner) (7), AugtyroTM (Repotrectinib) (8), OgsiveoTM (Nirogacestat) (9), WeliregTM (Belzutifan) (10), IwilfinTM (Eflornithine) (11), and ZoryveTM (Roflumilast) (12). Where it is possible, we will discuss the mode of biological activity, emphasizing the role of fluorine substitution in the development of a given drug. The total synthesis of these drugs and methods used for the introduction of fluorine will be discussed.

|
Download:
|
| Fig. 1. Structures of fluorine-containing drugs approved for the clinic in 2023. | |
{kind=link}
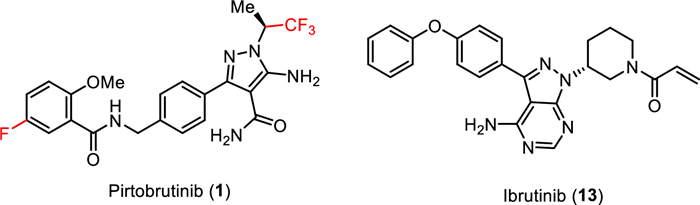
Pirtobrutinib (1) is a highly selective, non-covalent (reversible), orally bioavailable Bruton’s tyrosine kinase (BTK) inhibitor (Fig. 2) [43] developed by Eli Lilly. BTK plays a critical role in the development, activation, and survival of B cells [43]. B cells belong to the lymphocyte subtype of white cells and are responsible for producing antibodies, but uncontrolled growth of B cells can lead to cancer. Pirtobrutinib (1) inhibits BTK in a different manner compared to the previously approved BTK inhibitor ibrutinib (13) which covalently and irreversibly binds to the C481 residue in the kinase domain of BTK [44]. Therefore, pirtobrutinib (1) is effective for the treatment of ibrutinib-resistant chronic lymphocytic leukemia that develops due to C481 kinase domain mutations [45]. Chemically, pirtobrutinib (1) is based on the aminopyrazole carboxamide ring designed to replace the 4-aminopyrazolopyrimidine core of ibrutinib (13). The pyrazole ring serves as an ATP analog competitively binding to the ATP binding site of BTK and facilitating the proper conformation for effective inhibition. The covalent binding region of ibrutinib (13) was changed to a non-covalently bonded CF3-substituted ethyl group. The FDA approved pirtobrutinib (1) in January 2023 for the treatment of adult patients with relapsed or refractory mantle cell lymphoma (MCL) after at least two lines of systemic therapy, including a BTK inhibitor [46–48].

|
Download:
|
| Fig. 2. Structure of pirtobrutinib (1) and ibrutinib (13), a clinically approved BTK inhibitor. | |
{kind=link}
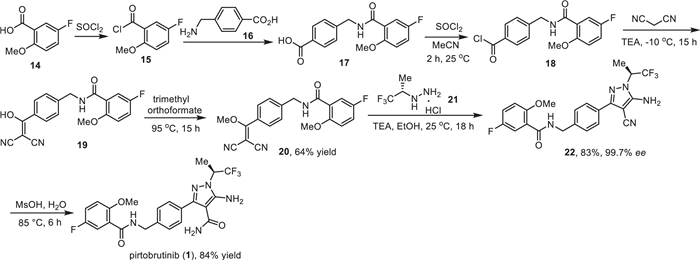
The synthesis of pirtobrutinib (1) developed by Loxo Oncology was shown in Scheme 1 [49]. Chlorination of 5-fluoro-2-methoxybenzoic acid (14) with thionyl chloride gave the corresponding acid chloride 15, which then reacted with 4-(aminomethyl)benzoic acid (16) to provide amido acid 17. Chlorination of amido acid 17 with thionyl chloride followed by condensation of the resulting acid chloride 18 without isolation with malononitrile in the presence of trimethylamine (TEA) in acetonitrile at −10 ℃ gave 2-(hydroxymethylene)malononitrile derivative 19. Subsequent O-methylation of 19 with trimethyl orthoformate at 95 ℃ led to 2-(methoxymethylene)malononitrile derivative 20 in 64% yield. Next, the cycloaddition reaction of compound 20 with (S)-(2,2,2-trifluoro-1-methyl)ethylhydrazine hydrochloride (21) in the presence of Et3N in EtOH provided 5-aminopyrazole-4-carbonitrile 22 in 83% yield and excellent enantiomeric purity (99.7% ee). The 5-aminopyrazole-4-carbonitrile 22 underwent hydrolysis by treatment with methanesulfonic acid (MsOH) in H2O at 55 ℃ to give pirtobrutinib (1) in 84% yield as a white solid.

|
Download:
|
| Scheme 1. Synthesis of pirtobrutinib (1). | |
{kind=link}
3. SkyclarysTM (Omaveloxolone)
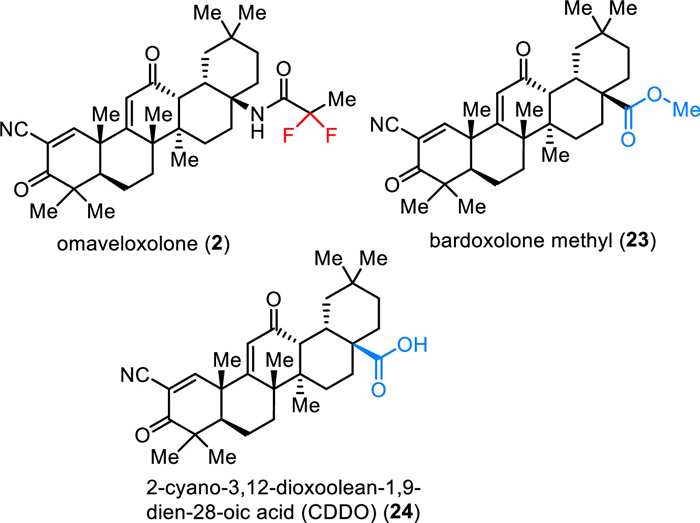
Omaveloxolone (2) is an orally active antioxidative transcription factor Nrf2 activator which was developed by Reata Pharmaceuticals Inc. (Fig. 3) [50]. Nrf2 is the principal regulator of the phase Ⅱ cellular antioxidant response and plays an important role in neuroprotection and detoxification [51]. Under physiological conditions, Nrf2 is sequestered in the cytosol by binding to protein Keap1 and is subsequently ubiquitinated and degraded [52]. Oxidative stress prevents ubiquitination and allows Nrf2 to translocate to the nucleus. Thus, this can activate the genes containing an antioxidant response element (ARE). Omaveloxolone (2), the related triterpenoids bardoxolone methyl 23 and 2-cyano-3,12-dioxoolean-1,9–dien-28-oic acid (CDDO) 24 bearing a cyanoenone moiety, which can covalently and reversibly bind to protein KEAP1 to inhibit ubiquitination and proteasomal degradation of Nrf2 [52,53]. Among triterpenoids, omaveloxolone (2) containing a 1,1-difluoroethylamide moiety was reported as one of the most potent compounds inducing ARE activity over 16-fold at 62.5 nmol/L in an Nrf2-GST-ARE luciferase reporter assay [54]. Omaveloxolone (2) was approved by the FDA in February 2023 as the first treatment for Friedreich’s ataxia, a rare inherited, degenerative disease that damages the nervous system, resulting in uncoordinated muscle movement, poor balance, difficulty walking, changes in speech and swallowing [55,56].

|
Download:
|
| Fig. 3. Structure of covalent Keap1 binders. | |
{kind=link}
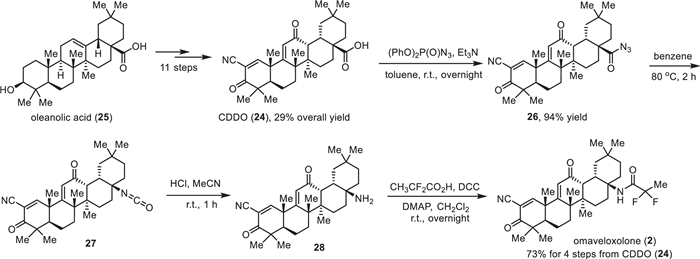
Synthesis of omaveloxolone (2) developed by Reata Pharmaceuticals was based on using CDDO (24) as the starting material (Scheme 2) [57]. CDDO (24), in turn, originated in 11 steps from the natural product oleanolic acid (25) with a 29% overall yield [58]. Conversion of carboxylic acid 24 using diphenylphosphoryl azide (DPPA) in the presence of triethylamine at room temperature provided the corresponding acyl azides 26 in 94% yield. Further Curtius rearrangement of acyl azide 26 by heating in anhydrous benzene afforded isocyanate 27, which was directly hydrolyzed by treatment with HCl in MeCM at room temperature to produce the corresponding amine 28 with retention of (R)-configuration. Omaveloxolone (2) was synthesized through peptidic coupling reactions between amine 28 and 2,2-difluoropropanoic acid. The reaction was performed using pre-activation of the carboxylic group with dicyclohexylcarbodiimide (DCC) and the desired omaveloxolone (2) was obtained in 73% yield from CDDO (24) over four steps.

|
Download:
|
| Scheme 2. Synthesis of omaveloxolone (2). | |
{kind=link}
4. JoenjaTM (Leniolisib)
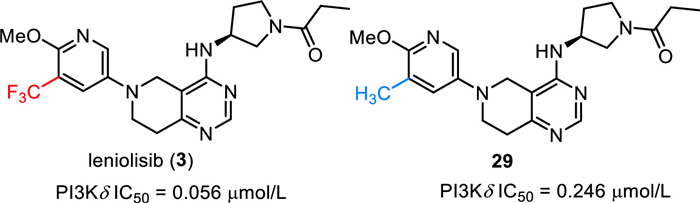
Leniolisib (3), also named CDZ173, was discovered by Novartis and developed by Pharming Group as a selective small molecule phosphoinositide 3-kinase-delta (PI3Kδ) inhibitor for the treatment of immunodeficiency disease [59–61]. As shown in Fig. 4, leniolisib (3) is a chiral compound, which features a key 5,6,7,8-tetrahydropyrido[4,3-d]-pyrimidine bicyclic fragment, a (S)−3-aminopyrrolidine moiety and a trifluoromethylated pyridine. Leniolisib (3) could suppress the hyperactivated PI3Kδ via selective inhibition of the p110δ subunit of PI3K. Leniolisib (3) showed good inhibitory activity against PI3K with the cellular IC50 of 1.67 µmol/L, 2.25 µmol/L and 0.056 µmol/L for PI3Kα, PI3Kβ, and PI3Kδ respectively. In particular, leniolisib (3) showed a good cellular selectivity against PI3Kδ with about thirty-fold over PI3Kα [59,62]. The structure-activity relationship (SAR) study by Novartis disclosed that the trifluoromethyl group played an important role in the activity. Compared with the non-fluorinated compound 29, introduction of a trifluoromethyl group to the methoxypyridine moiety led to a four-fold increased PI3Kδ potency (29, IC50 = 0.246 µmol/L) [59]. Furthermore, Leniolisib features acceptable hydrophilicity, good solubility, and metabolic stability, as well as favorable membrane permeability. Based on the findings from Phase Ⅱ/Ⅲ clinical trial, leniolisib (3) received its first approval by the FDA in March 2023 with the name JoenjaTM for use in the treatment of activated phosphoinositide 3-kinase delta syndrome (APDS) in adult and pediatric patients of 12 years of age and older [63].

|
Download:
|
| Fig. 4. Structure of leniolisib (3) and its analog. | |
{kind=link}
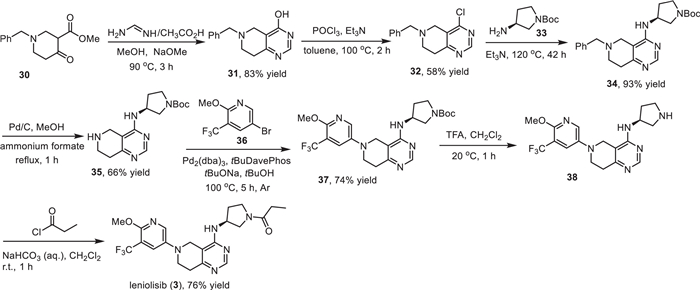
Novartis developed a synthetic strategy for the preparation of leniolisib (3), which used a tetrahydropyrido[4,3-d]pyrimidine heterocyclic compound 32 as the key intermediate (Scheme 3) [59,60,64,65]. First, the assembly of the fused pyrimidine cycle was conducted via a cyclization reaction of methyl 1-benzyl-4-oxopiperidine-3-carboxylate (30) with acetic acid methanimidamide in the presence of sodium methoxide. Then, the generated intermediate 31 was treated with phosphorus oxychloride (POCl3) with triethylamine as a base in toluene at 100 ℃ giving the chlorinated tetrahydropyrido[4,3-d]pyrimidine intermediate 32 in 58% yield [60]. Substitution reaction of the intermediate 32 by Boc-protected (S)−3-aminopyrrolidine 33 in a sealed vial at 120 ℃ delivered the chiral compound 34. This step introduced the desired chiral carbon center with (S)-configuration into leniolisib (3). Compound 34 underwent a Pd-catalyzed benzyl deprotection reaction in the presence of ammonium formate resulting in the fused heterocycle compound 35. Then, Pd-catalyzed coupling reaction of compound 35 with 5–bromo-2–methoxy-3-(trifluoromethyl)pyridine (36) by using 2-(di–tert-butylphosphino)−2′-(N,N-dimethylamino)biphenyl (tBuDavePhos) as a ligand successfully installed the trifluoromethylated pyridine into the fused heterocycle core. Finally, removing the Boc group of compound 37 via treatment with trifluoroacetic acid (TFA) in dichloromethane and followed by acylation with propionyl chloride afforded the desired leniolisib (3) in 76% yield [59].

|
Download:
|
| Scheme 3. Synthesis of leniolisib (3). | |
{kind=link}
5. MieboTM (Perfluorohexyloctane ophthalmic solution)
Perfluorohexyloctane (4), also named NOV03, was developed by Novaliq and Bausch + Lomb Corporation. It was used as an investigational, proprietary, water-free, single-component preservative-free eye drop to reduce tear evaporation at the ocular surface [66–70]. As shown in Fig. 1, perfluorohexyloctane (4) is a partially fluorinated linear alkane, which contains a perfluorohexyl moiety and an eight-carbon saturated alkyl species. Usually, perfluoroalkyl compound is regarded as an important component of fluorophors and the introduction of fluorine atoms can increase the lipophilicity of the molecules [71]. Perfluorohexyloctane could form a monolayer at the air-liquid interface of the tear film which led to the reducing of water evaporation. Novaliq designed a series of semifluorinated alkanes with different lengths of perfluoroalkyl chains and saturated alkyl chains. The related SAR studies led to the discovery of perfluorohexyloctane (4) [67,68]. Based on the results of the phase trial, perfluorohexyloctane ophthalmic solution was approved by the FDA in May 2023 with the trade name MieboTM specifically for the treatment of the signs and symptoms of dry eye disease.
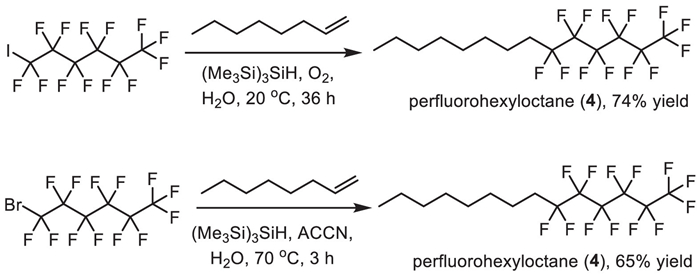
The synthesis of perfluorohexyloctane (4) was shown in Scheme 4. Its synthesis in organic solvent could be achieved via a radical addition of perfluoroalkyl radicals to carbon-carbon unsaturated bonds. In 2010, Postigo and co-authors reported a (Me3Si)3SiH-promoted radical perfluoroalkylation reaction of oct–1-ene with different perfluoroalkyl sources [72]. Using 1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluoro-6-iodohexane as a source in the presence of oxygen at 20 ℃ generated perfluorohexyloctane (4) with 74% yield after 36 h. Also, 1,1,1,2,2,3,3,4,4,5,5,6,6-tridecafluoro-6-bromohexane was used as substrate in the presence of 1,1′-azobis(cyclohexanecarbonitrile) (ACCN) at 70 ℃ afforded the desired perfluorohexyloctane (4) in 65% yield after 3 h (Scheme 4).

|
Download:
|
| Scheme 4. Synthesis of perfluorohexyloctane (4). | |
{kind=link}
6. VeozahTM (Fezolinetant)
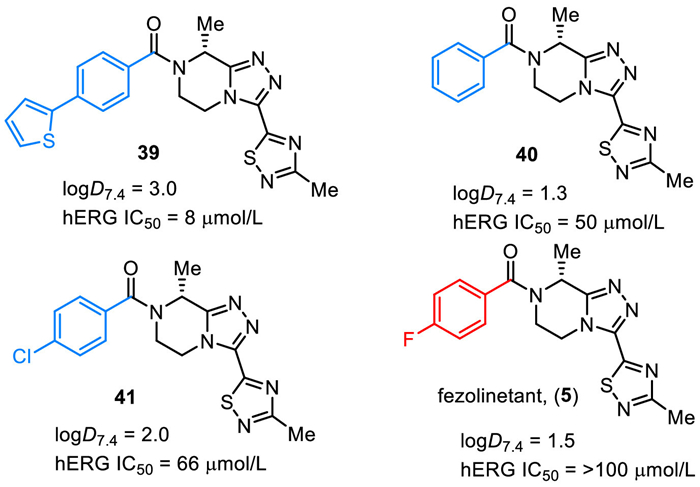
Fezolinetant (5) was discovered by Euroscreen S.A. as an oral, small molecule, neurokinin 3 receptor (NK3R) antagonist for the treatment of sex-hormone disorders [73–75]. Fezolinetant (5) is a chiral compound featuring a triazolopiperazine core, a 1,2,4-thiadiazole unit, and a 4-fluorobenzoyl group (Fig. 5). The in vitro SAR studies disclosed that fluorine substituent on the phenyl ring plays an important role, and fezolinetant (5) was the best one overall in the hERG (human ether-a-go-go-related gene) and CYP (cytochrome P450) safety profile. For example, the logD7.4 was 3.0 and hERG IC50 was 8 µmol/L for compound 39. Replacing the thienyl substituent by a fluoro group resulted in reduced lipophilicity (ΔlogD7.4 = −1.5) and improved over 12-fold hERG IC50 (fezolinetant (5), logD7.4 = 1.5, hERG IC50 = > 100 µmol/L). Also, the compounds with no substituent (40) and chloro (41) on phenyl were evaluated and no improved hERG IC50 was observed compared with fezolinetant (5) [74,75]. Fezolinetant is being developed by Astellas Pharma Inc. and currently has emerged as a novel therapeutic strategy. Fezolinetant can obviously reduce the frequency and severity of vasomotor symptoms (VMS) and shows fewer side effects. In May 2023, fezolinetant (VeozahTM) received its first approval by the FDA as a first-in-class non-hormonal treatment of moderate to severe VMS or menopausal hot flashes [76,77].

|
Download:
|
| Fig. 5. Structures of fezolinetant (5) and its analogs. | |
{kind=link}
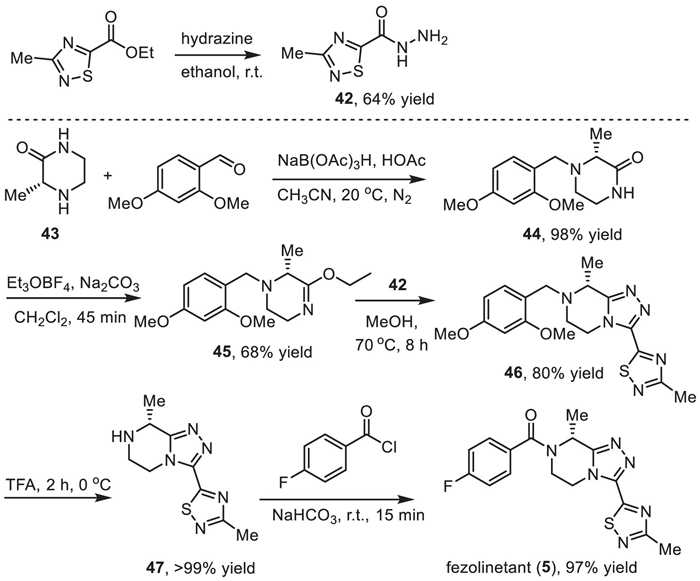
The preparative route for the synthesis of fezolinetant (5) developed by Euroscreen S.A. was illustrated in Scheme 5 with the formation of the fused triazolopiperazine 46 via cyclodehydration of piperazinoimidate and 1,2,4-thiadiazole-5-carbohydrazide 42 as a key step [74,75]. The desired (R)-carbon center in fezolinetan (5) was introduced via the use of (R)−3-methylpiperazin-2-one (43) as the starting material. The 1,2,4-thiadiazole-5-carbohydrazide intermediate 42 was obtained in 64% yield via the reaction between ethyl 3-methyl-1,2,4-thiadiazole-5-carboxylate and hydrazine in ethanol at room temperature. On the other hand, reduction amination of (R)−3-methylpiperazin-2-one (43) and 2,4-dimethoxybenzaldehyde with sodium triacetoxyborohydride as a reductive reagent in the presence of acetic acid under nitrogen atmosphere afforded the 2,4-dimethoxybenzyl protected intermediate 44 [78]. By using Meerwein reagent (Et3OBF4) and a mild base Na2CO3, the piperazin-2-one 44 was converted into the piperazinoimidate intermediate 45 in 68% yield and 98.8% ee. It should be mentioned that the use of buffered Meerwein conditions could minimize racemization during this step [74]. Subsequently, the piperazinoimidate underwent a cyclodehydration reaction with the 1,2,4-thiadiazole-5-carbohydrazide intermediate 42 at 70 ℃ for 8 h giving the fused triazolopiperazine 46 in 80% yield. Removal of the protecting 2,4-dimethoxybenzyl group via the treatment with TFA at 0 ℃ gave the free amine 47 in > 99% yield, which was used for the reaction with 4-fluorobenzoyl chloride at room temperature to achieve the formation of fezolinetan (5) in 97% yield.

|
Download:
|
| Scheme 5. Synthesis of fezolinetan (4). | |
{kind=link}
7. PaxlovidTM (Nirmatrelvir, ritonavir)
Paxlovid is an oral antiviral drug containing nirmatrelvir (6A) and ritonavir (6B). It was developed by Pfizer and received FDA approval in May 2023 for the treatment of mild-to-moderate cases of coronavirus disease in patients who are at high risk for progression to severe COVID-19 [79]. Nirmatrelvir (6A), originally called PF-07321332, as a protease inhibitor, disrupts the viral life cycle by targeting the SARS-CoV-2 main protease (MPro). This inhibition is crucial for preventing the virus from multiplying. Importantly, the low off-target activity of nirmatrelvir (6A) is beneficial for reducing potential side effects associated with affecting human proteins [80,81]. Ritonavir (6B), in this combination treatment, serves a dual purpose. First, as a human HIV protease inhibitor, it contributes to the antiviral effect. Second, ritonavir (6B) acts as a pharmacokinetic enhancer by inhibiting CYP3A4, an enzyme involved in drug metabolism. By slowing down the metabolism of nirmatrelvir, ritonavir helps to increase its bioavailability and prolong its presence in the body. This mechanism allows for higher concentrations of nirmatrelvir, enhancing its efficacy in combating the SARS-CoV-2 virus [82,83]. The combination of nirmatrelvir and ritonavir, as described, illustrates a targeted and synergistic approach to inhibiting viral replication and treating early COVID-19 infections, with the potential to prevent the progression to more severe symptoms.
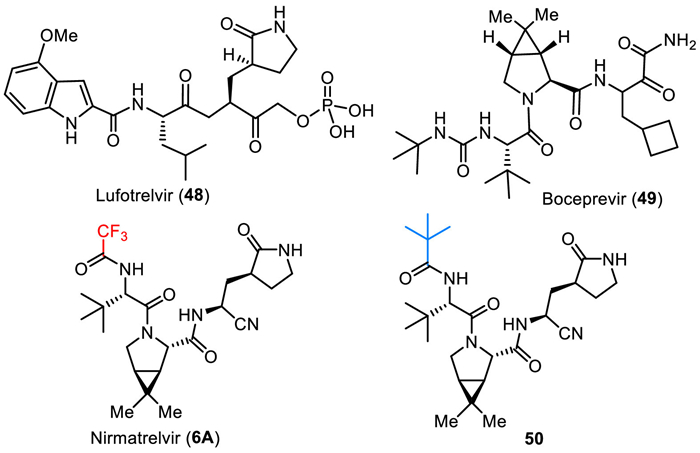
Nirmatrelvir (6A) is described as a protein mimetic tripeptide, and it evolved from SAR studies on lufotrelvir (48) and a dipeptide of boceprevir (49) (Fig. 6) [84]. The pyrrolidine amino acid fragment from lufotrelvir (48) and the dipeptide fragment from boceprevir (49) contributed to the foundational structure of nirmatrelvir. The phosphate prodrug functionality present in lufotrelvir was replaced with a cyano group in nirmatrelvir. This modification likely affects the compound’s pharmacokinetics and bioavailability. Exploratory studies for boceprevir revealed that the incorporation of the gem–dimethyl cyclopropyl proline moiety increased the potency of the molecule. This finding was likely influential in the design of nirmatrelvir for treating SARS-CoV-2. The strategy of incorporating a bicyclic amino acid, similar to what was used in boceprevir exploratory studies, was employed to identify potent molecules for treating SARS-CoV-2. This suggests a rational design approach based on known structural features that enhance antiviral activity. Also, nirmatrelvir (6A) features a trifluoromethyl group, which is important for the bioactivity disclosed by the SAR studies. The enzymatic IC50, cellular EC50, and antiviral EC50 values of MPro inhibitory activity for nirmatrelvir (6A) were 66 nmol/L, 3.4 µmol/L, and 1.3 µmol/L, respectively. However, changing trifluoromethyl to tert–butyl (50) led to an eleven-fold decrease in inhibitory activity (enzymatic IC50 = 720 nmol/L) [85].

|
Download:
|
| Fig. 6. Structures of lufotrelvir (48), boceprevir (49), nirmatrelvir (6A) and its analog 50. | |
{kind=link}
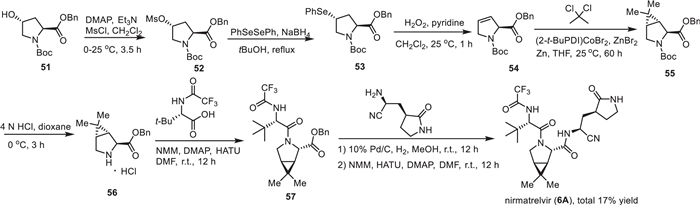
The synthesis of nirmatrelvir (6A) and several other analogs was described in patents of Pfizer [86,87]. The synthesis started with the conversion of Boc-protected trans-4–hydroxy-l-proline benzylester 51 to methanesulfonate 52 with the use of methanesulfonyl chloride (MsCl), N,N-dimethylpyridin-4-amine (DMAP) and trimethylamine. Methanesulfonate 52 reacted with PhSeSePh generating 4-(phenylselanyl)pyrrolidine 53 under reflux. Then, oxidation of 4-(phenylselanyl)pyrrolidine 53 followed by elimination of the hydroxy group delivered the alkene intermediate 54. The resulting alkene 54 underwent a cyclopropanation reaction with 2,2-dichloropropane catalyzed by (2-t-BuPDI)CoBr2 in the presence of ZnBr2 and Zn yielding a bicyclic compound 55 that was then converted to compound 56 after removal of Boc protecting group under acidic conditions. Subsequently, coupling of 56 with (S)−3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoic acid by using 2-(7-azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU) in the presence of 4-methylmorpholine (NMM) and DMAP afforded CF3-containg compound 57. Deprotection of the benzyl group of ester 57 and followed by coupling with (S)−2-amino-3-((S)−2-oxopyrrolidin-3-yl)propanenitrile resulted in nirmatrelvir (6A) with approximately total 17% yield for the multistep synthesis (Scheme 6) [88].

|
Download:
|
| Scheme 6. Synthesis of nirmatrelvir (6A). | |
{kind=link}
8. XdemvyTM (Lotilaner)
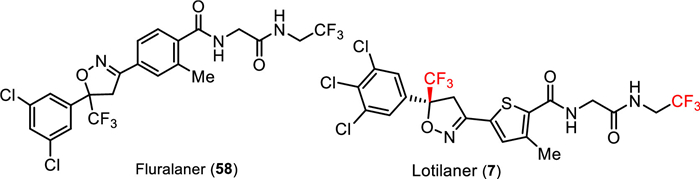
A series of reported antiparasiticides contain the isoxazoline moiety such as fluralaner, afoxolaner, and sarolaner [89,90]. Several research groups have reported that isoxazoline derivatives can act as specific blockers of γ-aminobutyric acid gated chloride (GABACl) ion channels and glutamate-gated chloride (GluCl) channels in insects [91]. Isoxazolines, including lotilaner, have been widely as veterinary medicines for the treatment of flea and tick infestations in animals. Lotilaner (7), developed by Tarsus Pharmaceuticals Inc. and received FDA approval in July 2023, is an antiparasitic agent (ectoparasiticide) for the treatment of Demodex blepharitis. Demodex mites that cause blepharitis are the ectoparasites which mostly live in hair follicles and oil glands on the face, neck, or chest in humans. Blepharitis is an inflammatory disease of the eyelid that causes redness and eye irritation. Besides Demodex blepharitis, lotilaner could also be used for the control of flea allergy dermatitis (FAD) [92–95]. Credelio Plus is a combination of lotilaner and milbemycin oxime used for the treatment of parasitic infections in animals [96,97].
Lotilaner (7) acts as a non-competitive antagonist of GABACl, selectively targeting mites without inhibiting mammalian GABA-mediated chloride channels at the recommended human ophthalmic dose as it does not bind to the same therapeutic target in mammals [98]. Similar to fluralaner (58), lotilaner contains a chiral center with two enantiomers (Fig. 7). Only the S-enantiomer of lotilaner (7) is active, while the R-enantiomer exhibits low biological activity. Inhibition of GABACl by lotilaner (7) causes a paralytic action in the target organism, leading to death. This mechanism is effective for treating conditions such as blepharitis, meibomian gland dysfunction, and Lyme disease [99]. Investigations were conducted to explore if the mode of action and binding sites of lotilaner (7) were similar to GABACl blockers like dieldrin or fipronil.

|
Download:
|
| Fig. 7. Structures of chloride channel antagonists fluralaner (58) and lotilaner (7). | |
{kind=link}
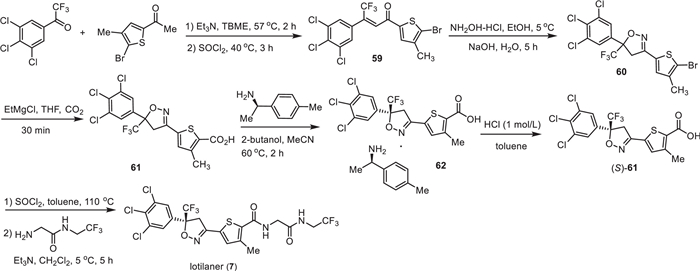
The preparation of lotilaner (7) used 2,2,2-trifluoro-1-(3,4,5-trichlorophenyl)ethan-1-one as the starting material, which reacted with 1-(5–bromo-4-methylthiophen-2-yl)ethan-1-one with tert–butyl methyl ether (TBME) as solvent in the presence of Et3N to give the intermediate 59 [100]. Then, compound 59 underwent a cyclization reaction with NH2OH in the presence of NaOH leading to the formation of isoxazole intermediate 60. The carboxylic group was introduced via reaction with EtMgCl in THF under a carbon dioxide atmosphere. Chiral separation was used via the formation of salt 62 by using (R)−1-(p-tolyl)ethan-1-amine, which was treated with HCl to provide the chiral intermediate 61 with (S)-configuration. Finally, the acid 61 was converted into acyl chloride and then directly reacted with 2-amino-N-(2,2,2-trifluoroethyl)acetamide in the presence of trimethylamine affording the corresponding lotilaner (7) (Scheme 7).

|
Download:
|
| Scheme 7. Synthesis of lotilaner (7). | |
{kind=link}
9. AugtyroTM (Repotrectinib)
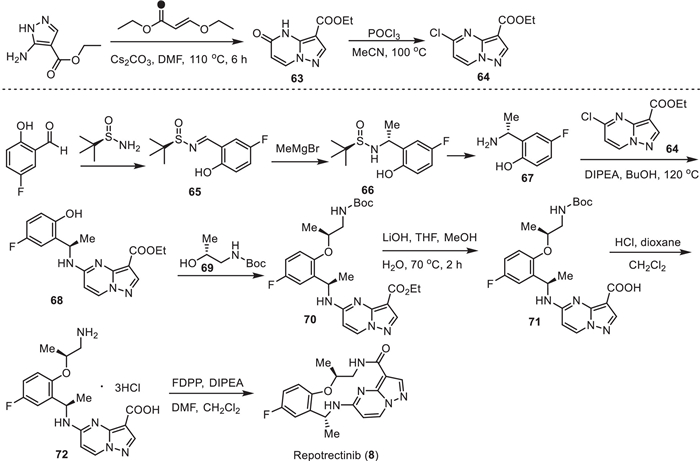
Repotrectinib (TPX-0005) (8) (Fig. 1) is a next-generation tyrosine kinase inhibitor (TRKI). It shows high inhibitory activity against proto-oncogene tyrosine-protein kinase ROS (ROS1) and the tropomyosin receptor tyrosine kinases (TRKs) TRKA, TRKB, and TRKC. Repotrectinib (8) is an anti-cancer drug and is used for the treatment of non-small cell lung cancer. Repotrectinib (8) was approved by the FDA for medical use in the United States in November 2023 under the brand name Augtyro [101,102]. Structurally, repotrectinib (8) is a macrocyclic lactam featuring fused heteroaromatic and benzene rings connected via amino and 1,2-hydroxyamino residues bearing two stereogenic carbons. Fluorination in repotrectinib (8) is represented by a single aromatic fluorine, located in para-position to the ether oxygen atom. This pattern of fluorination is commonly used to prevent oxidative metabolic degradation of aromatic moieties [26].
One of the key intermediates in the preparation of repotrectinib (8) is chloropyrazolo[1,5-a]pyrimidine 64 (Scheme 8). The procedure started with the condensation of ethyl 5-amino-1H-pyrazole-4-carboxylate with (E)-ethyl 3-ethoxyacrylate to furnish ethyl 4,5-dihydro-5-oxopyrazolo[1,5-a]pyrimidine-3-carboxylate (63). Its conversion to 64 via chlorination aromatization was performed using POCl3 in MeOH at 100 ℃ [103,104].

|
Download:
|
| Scheme 8. Synthesis of repotrectinib (8). | |
{kind=link}
The second key intermediate in the synthesis of repotrectinib (8) is 2-((R)−1-aminoethyl)−4-fluorophenol (67) (Scheme 8). A convenient approach for its synthesis involves the application of tert–butanesulfinamide [105,106] as a chiral auxiliary. Sulfinyl imine 65, prepared from 5-fluoro-2-hydroxybenzaldehyde and tert–butanesulfinamide, reacted with MeMgBr to furnish addition product 66 with excellent diastereoselectivity (> 95% de). Purification and deprotection of intermediate 66 under usual acidic conditions [107] gave the target free amine 67 in enantiomerically pure form. Then, aromatic nucleophilic substitution of the chlorine atom in 64 by the amino group in 67 under forcing condition at 129 ℃ using DIPEA [108] as a base [103,104] afforded compound 68. The resulting compound 68 was further reacted with commercially available tert–butyl (R)-(2-hydroxypropyl)carbamate (69) to form the corresponding ester 70. Deprotection of the ester faction in 70 and N-Boc group in product 71, under the correspondingly basic and acidic conditions, gave rise to product 72 as a triple hydrochloric salt. Finally, cyclization of compound 72 via a peptide bond-forming reaction using pentafluorophenyl diphenylphosphinate (FDPP) as a coupling agent provided the desired repotrectinib (8) [109].
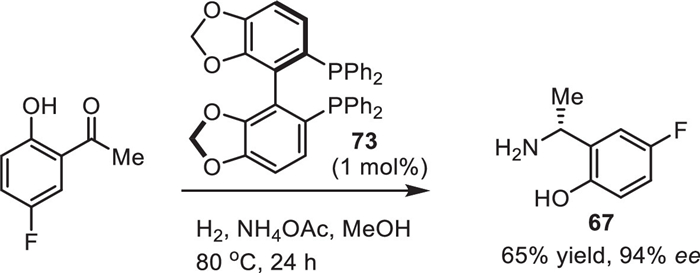
Here we would like to mention another approach to the intermediate 67 via a catalytic enantioselective reductive amination [110]. As shown in Scheme 9, the target product (R)−67 was prepared from 1-(5-fluoro-2-hydroxyphenyl)ethan-1-one using Ru-chelated phosphine ligand 73 as a catalyst and NH4OAc a source of nitrogen and H2 as a reducing reagent. The reaction was reproduced on a 20 g scale with respectable stereochemical outcome. Unfortunately, the claimed enantioselectivity was not undoubtedly confirmed by the necessary in such cases SDE study [111]. One could take a note that fluorine-containing chiral amines [112], and α-phenylethylamines [113] usually show quite an exceptional magnitude of SDE under the conditions of achiral column chromatography.

|
Download:
|
| Scheme 9. Catalytic enantioselective reductive amination for the synthesis of amine 67. | |
{kind=link}
10. OgsiveoTM (Nirogacestat)
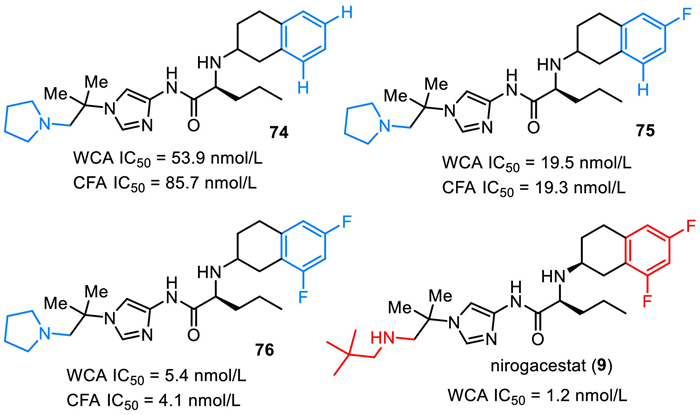
Nirogacestat (9), also named PF-3084014, was discovered and reported by Pfizer Phama in 2011 [114]. Nirogacestat was developed as an γ-secretase inhibitor that blocks proteolytic activation of the Notch receptor [114–117]. As shown in Fig. 8, nirogacestat is a chiral compound with two (S)-carbon centers. It contains three key structural units, including difluorinated tetralin, L-norvaline, and amino imidazole species. Careful SAR studies disclose that the fluoro substituent plays a key role in the activity of reducing Aβ. Compared with non-fluorinated compound 74 (CFA IC50 = 85.7 nmol/L), introducing one fluorine atom at the tetralin ring led to about five-fold potency improvement in the cell free assay (compound 75, CFA IC50 = 19.3 nmol/L). Notably, compound 76 featuring a difluorinated tetralin unit showed further improved activity with WCA IC50 value of 5.4 nmol/L and CFA IC50 value of 4.1 nmol/L [114]. Further SAR studies via replacing the pyrollidine species by 2,2-dimethylpropan-1-amine brought a better activity (WCA IC50 = 1.2 nmol/L), which was thus selected for clinical development. Nirogacestat (9) was developed by SpringWorks Therapeutics, and got its approval by the FDA with the trade name OgsiveoTM in November 2023 for the treatment of desmoid tumors based on results from the Phase Ⅲ DeFi trial. In particular, nirogacestat (9) was used for adults with progressing desmoid tumors who require systemic treatment.

|
Download:
|
| Fig. 8. Structures of nirogacestat (9) and its analogs. | |
{kind=link}
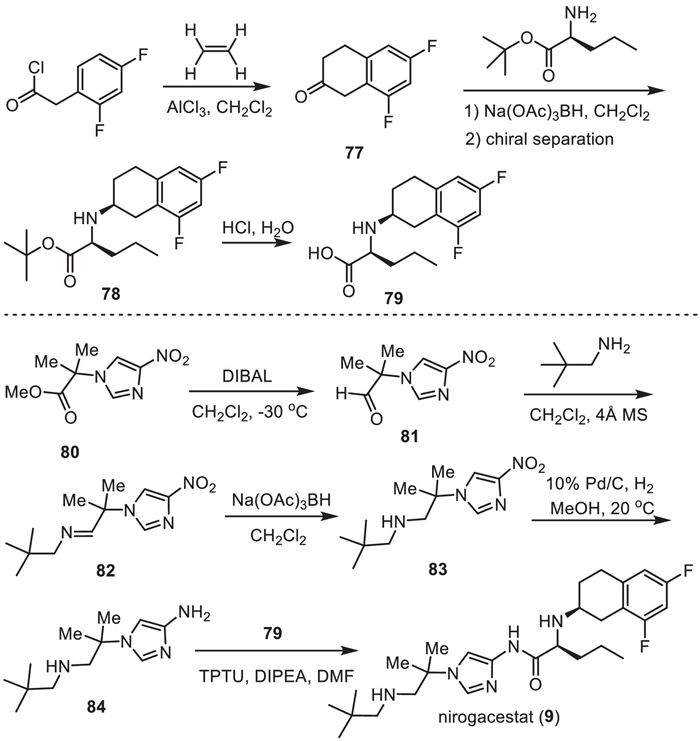
Pfizer Phama developed a synthetic method for the preparation of nirogacestat (9), which used chiral fluorinated tetralin-containing amino acid 79 and amino imidazole 84 as the two key intermediates (Scheme 10) [114,118]. Friedel-Crafts acylation of 2,4-difluorophenyl acetyl chloride with ethylene in the presence of AlCl3 in dichloromethane afforded 6,8-difluoro-3,4-dihydronaphthalen-2(1H)-one (77). Then, reduction amination of the ketone 77 with L-norvaline t–butyl ester by using Na(OAc)3BH as a reductive reagent generated a mixture of racemic 78, which was subjected to chiral HPLC separation affording the chiral 78 with (S,S) absolute confirmation. After treatment by hydrochloride, the t–butyl ester was converted into amino acid 79.

|
Download:
|
| Scheme 10. Synthesis of nirogacestat (9). | |
{kind=link}
Another intermediate 84 was synthesized starting with nitroimidazole 80. Reduction of the ester group of nitroimidazole 80 by diisobutyl aluminium hydride (DIBAL) at −30 ℃ provided the corresponding aldehyde 81, which underwent a condensation reaction with 2,2-dimethylpropan-1-amine in the presence of 4 Å MS to give the imine 82. Imine 82 was then reduced by Na(OAc)3BH delivering nitroimidazole featuring secondary amino group 83. Subsequently, hydrogenation of the nitroimidazole 83 with 10% Pd/C as a catalyst in methanol at 20 ℃ provided the aminoimidazole intermediate 84. Finally, condensation reaction of amino acid 79 and aminoimidazole intermediate 84 in the presence of O-(1,2-dihydro-2-oxo-pyridyl)−1,1,3,3-tetramethyluronium tetrafluoroborate (TPTU), DIPEA to furnish the desired nirogacestat (9).
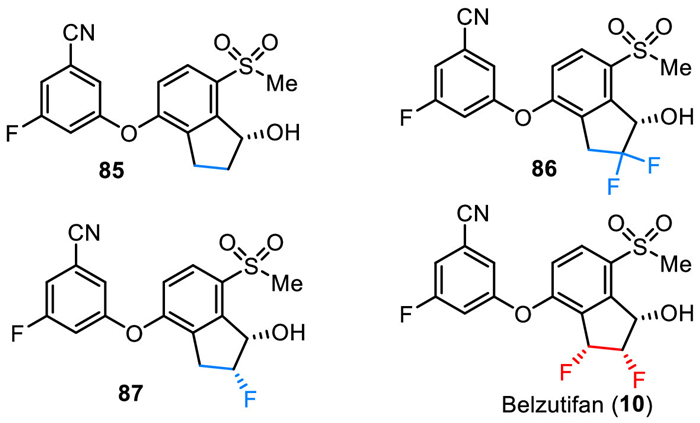
11. WeliregTM (Belzutifan)Belzutifan (PT2977) was developed by Peloton Therapeutics and Merck & Co. as a small molecule hypoxia-inducible factor (HIF)−2α inhibitor for the treatment of solid tumors, including clear cell renal cell carcinoma (ccRCC) and von Hippel–Lindau (VHL) disease-associated RCC [119–122]. As shown in Fig. 9, belzutifan (10) is a chiral molecule containing three continuous chiral carbon centers with (1S,2S,3R) configuration. It is a diaryl ether with a fluorinated phenyl moiety and a difluorinated indanol fragment. The SAR studies via testing in the SPA binding assay and HIF-2α luciferase assay in 786-O cells by Peloton Therapeutics have disclosed that the inhibitory activity of belzutifan is much better than its non-fluorinated analogs [122]. For example, compound 85 with no fluorine atom on the indanol ring showed the SPA IC50 value of 0.16 µmol/L and the luciferase EC50 of 0.93 µmol/L. Compound 86 with a gem–difluoro moiety showed a 13-fold increase in potency (SPA IC50 = 0.012 µmol/L). Furthermore, changing the germinal difluoro group to the vicinal difluoro group led to a 17-fold increase in potency in SPA assay and a more than 80-fold increase in potency in luciferase assay (belzutifan (10), SPA IC50 = 0.009 µmol/L, luciferase EC50 of 0.011 µmol/L). In December 2023, belzutifan (WeliregTM) was approved by the FDA for the treatment of advanced renal cell carcinoma. It should be mentioned that it received its first approval from the FDA in August 2021 for the treatment of patients with certain types of VHL disease-associated tumors [123].

|
Download:
|
| Fig. 9. Structures of belzutifan (10) and its analogs. | |
{kind=link}
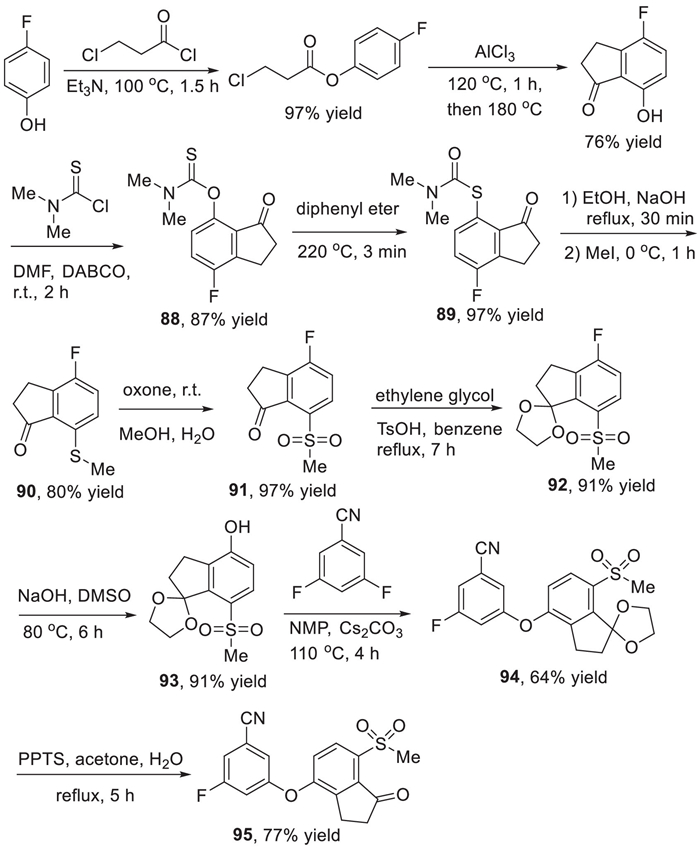
The synthesis of belzutifan (10) was presented in Scheme 11, which used 3-fluoro-5-((7-(methylsulfonyl)−1-oxo-2,3-dihydro-1H-inden-4-yl)oxy)benzonitrile (95) as a key intermediate and involved construction of the continuous three chiral carbon centers as the key steps [124]. The synthesis of the key diaryl ether intermediate 95 started from the acylation of 4-fluorophenol with 3-chloropropanoyl chloride in the presence of trimethylamine. The resulting ester was heated with AlCl3 via a Friedel-Crafts reaction generating the 4-fluoro-7–hydroxy indanone. The indanone reacted with N,N-dimethylthiocarbamoyl chloride with 1,4-diazabicyclo[2.2.2]octane (DABCO) as a base leading to the formation O-arylcarbamate 88 in 87% yield, which underwent a Newman-Kwart rearrangement affording the aryl shift compound 89 in 97% yield. Then, hydrolysis of the intermediate 89 under the basic conditions followed by methylation with MeI as a methyl source gave the intermediate 90 in 80% yield. Subsequently, oxidation of indanone 90 by using oxone as an oxidant at room temperature produced the sulfone intermediate 91 in 97% yield. Then, the ketone group of compound 91 was protected by ethylene glycol in the presence of TsOH, and the resulting ketal 92 underwent a SNAr substitution reaction by sodium hydroxide at 80 ℃ to provide the ketal phenol 93 in 91% yield. Ketal phenol 93 underwent a substitution reaction with 3,5-difluorobenzonitrile in the presence of Cs2CO3 and N-methylpyrrolidone (NMP) affording the desired diaryl ether intermediate 94 in 64% yield. The intermediate 94 was treated with pyridinium p-toluenesulfonate (PPTS) for 5 h affording the key intermediate 95 in 77% yield.

|
Download:
|
| Scheme 11. Synthesis of the intermediate 95. | |
{kind=link}
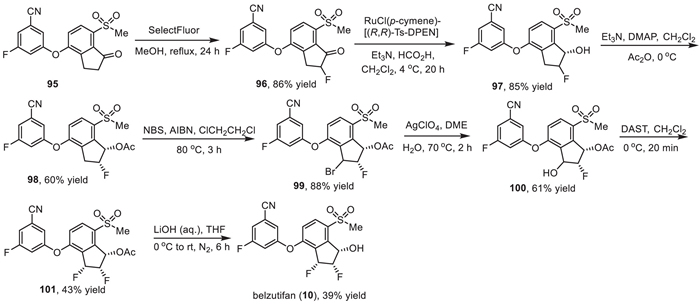
Then, the continuous chiral centers were installed on the diaryl ether intermediate 95 (Scheme 12). Fluorination of α-position of ketone via refluxing with SelectFluor in methanol afforded compound 96 in 86% yield. Asymmetric hydrogenation of compound 96 gave alcohol 97, which reacted with acetic anhydride generating the corresponding protected acetate 98 in cis- and trans-diastereomers. The cis- and trans-diastereomers could be separated by silica gel column resulting in the pure cis-isomer (1S, 2R)−98 in 60% yield. Subsequently, benzylic bromination of compound 98 by N-bromosuccinimide (NBS) initiated by 2,2′-azobis(2-methylpropionitrile) (AIBN) at 80 ℃ provided compound 99. Treatment of compound 99 with AgClO4 in 1,2-dimethoxyethane (DME) and water at 70 ℃ produced the alcohol 100. Then, alcohol 100 was subjected to a deoxyfluorination with N,N-diethylaminosulfur trifluoride (DAST) in dichloromethane resulting in the difluoro compounds with cis- and trans-diastereomers, which could be separated by silica gel column to successfully construct the three continuous chiral center with (1S,2S,3R) configuration (101) in 43% yield. Finally, deprotection of the acetyl group in the presence of aqueous LiOH furnished the desired belzutifan (10) in a 39% yield.

|
Download:
|
| Scheme 12. Synthesis of belzutifan (10). | |
{kind=link}
12. IwilfinTM (Eflornithine)
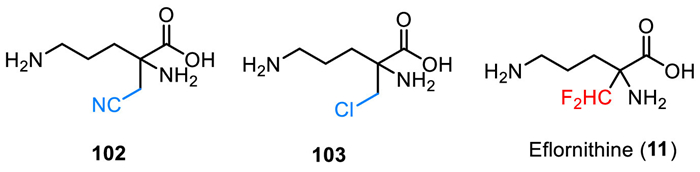
Eflornithine (α-difluoromethylornithine, DFMO, MDL-71728, IwilfinTM) was developed by US WorldMeds as an ornithine decarboxylase inhibitor [125,126], which was used specifically to reduce the relapse risk in adult and pediatric patients with high-risk neuroblastoma (HRNB) who have demonstrated at least a partial response to prior therapy. Eflornithine (11) is a white to off-white, odorless solid, which contains an amino acid fragment and a key difluoromethyl moiety at the α-position (Fig. 10). The chemistry structure of eflornithine (11) contains a chiral carbon center, however, it was used as a racemic mixture with d/l-isomers. Notably, Ashton and co-authors reported a chiral chromatographic method for the isolation of pure chiral isomer on a semi-preparative milligram scale [127]. The bioactive studies on catalytic irreversible inhibition of Mammalian ornithine decarboxylase (ODC) disclosed that the difluoromethyl group is essential for the inhibition process [128]. Eflornithine (11) showed the most efficient inhibitory activity compared with the other analogs featuring cyano or chloro substituent (102 and 103). Eflornithine (11) received its approval by the FDA in December 2023 for the treatment of high-risk neuroblastoma. It should be mentioned that eflornithine also has been used for the treatment of human African trypanosomiasis (HAT) caused by the parasite Trypanosoma brucei [129–131]. Also, eflornithine was approved by the FDA for the treatment of gambiense sleeping sickness in 1990 [129].

|
Download:
|
| Fig. 10. Structures of eflornithine (11) and its analogs. | |
{kind=link}
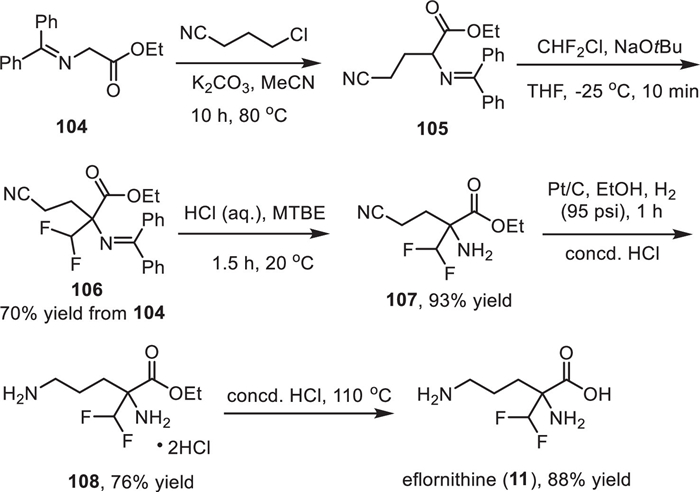
The synthetic route for the preparation of eflornithine (11) was presented in Scheme 13, which used N-(diphenylmethylene)glycine ethyl ester 104 as the starting material [132]. First, a substitution reaction between N-(diphenylmethylene)glycine ethyl ester and 3-chloropropionitrile with potassium carbonate as a base in acetonitrile at 80 ℃ afforded α-cyanoalkyl glycine ester 105. Then, a difluoromethyl group was installed to compound 105 via reaction with Freon-22 in the presence of NaOtBu with 70% yield of the two steps. Subsequently, treatment of the intermediate 106 with aqueous HCl in methyl tert–butyl ether (MTBE) provided the free amine 107 in 93% yield, which was subjected to a Pt/C-catalyzed reduction in which the cyano group was transferred into the amino group. Finally, hydrolysis of the ester group of 108 under acidic conditions at 110 ℃ provided the desired eflornithine (11) in 88% yield.

|
Download:
|
| Scheme 13. Synthesis of eflornithine (11). | |
{kind=link}
13. ZoryveTM (Roflumilast)
Roflumilast (APTA 2217, B9302–107, BY 217, BYK 20869) was developed by Altana Pharma (formerly Byk Gulden) as a selective phosphodiesterase 4 (PDE4) inhibitor [133–136]. It is an anti-inflammatory molecule and is used for the treatment of chronic obstructive pulmonary disease (COPD) [137–139]. The chemical name of roflumilast (12) is 3-(cyclopropylmethoxy)-N-(3,5-dichloropyridin-4-yl)−4-(difluoromethoxy)benzamide. It contains a benzamide fragment, a pyridine moiety, and a difluoromethoxyl group. BYK Gulden carried out the SAR studies and a series of fluoroalkoxy-substituted benzamides were prepared for the evaluation of phosphodiesterase 4 inhibition activity. The results led to the compound of roflumilast (12) [140].
In February 2011, roflumilast (DalirespTM) received its approval from the FDA for the treatment of chronic obstructive [133]. Also, roflumilast (12) with the trade name Zoryve was approved by the FDA in July 2022 by the company Arcutis Biotherapeutics for the treatment of plaque psoriasis in patients 6 years of age or older. In December 2023, Zoryve foam was approved by the FDA again for the treatment of seborrheic dermatitis in individuals 9 years of age and older.
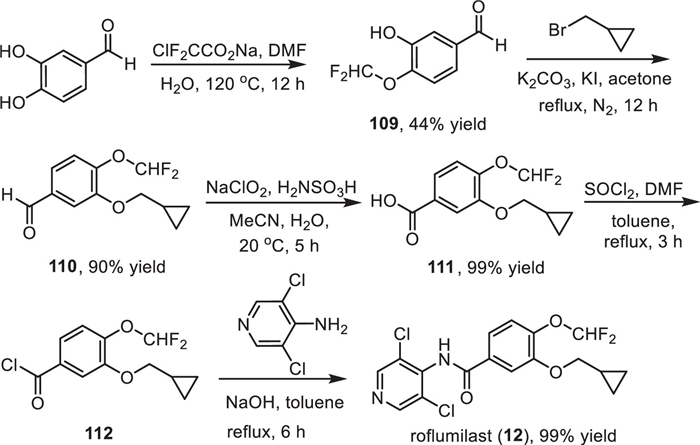
The synthesis of roflumilast (12) has been reported by several groups [141–143], and one convenient method for its preparation was presented in Scheme 14 with 4-difluoromethoxy-3-hydroxybenzaldehyde (109) as the starting material [144]. The difluoromethoxy substituted benzaldehyde 109 was obtained from a direct difluoromethylation reaction of 3,4-dihydroxybenzaldehyde with sodium chlorodifluoroacetate at 120 ℃ [145]. Then, a cyclopropylmethyl was introduced to another hydroxyl group via the reaction of the intermediate 109 with (bromomethyl)cyclopropane in the presence of K2CO3 and KI under reflux for 12 h. Subsequently, treatment of aldehyde 110 by sodium chlorite for 5 h afforded benzoic acid 111 in 99% yield. Then, reflux of benzoic acid 111 with SOCl2 in toluene for 3 h afforded the benzoyl chloride 112, which was directly subjected to the reaction with 3,5-dichloropyridin-4-amine under the basic conditions generating the desired roflumilast (12) in 99% yield.

|
Download:
|
| Scheme 14. Synthesis of roflumilast (12). | |
{kind=link}
14. Conclusions and outlook
In this year, the FDA approved fifty-five new drugs, among which there were thirty-two small-molecule drugs. In particular, twelve small molecules featuring various types of fluorinations were approved this year. These include JaypricaTM (Pirtobrutinib) (1), SkyclarysTM (Omaveloxolone) (2), JoenjaTM (Leniolisib) (3), MieboTM (Perfluorohexyloctane) (4), VeozahTM (Fezolinetant) (5), PaxlovidTM (Nirmatrelvir and Ritonavir) (6), XdemvyTM (Lotilaner) (7), AugtyroTM (Repotrectinib) (8), OgsiveoTM (Nirogacestat) (9), WeliregTM (Belzutifan) (10), IwilfinTM (Eflornithine) (11), and ZoryveTM (Roflumilast) (12). These drugs represent such therapeutic areas as cancer (1, 8, 9), neuromuscular disorder (2, 5), immunodeficiency (3), virology (6) and infectious diseases (7). As compared with the previous years, one can notice an increase in a relative proportion of molecules bearing multiple fluorine atoms (1–4, 6, 7, 9–12), while compounds 5 and 8 feature a most common type of a single aromatic fluorine. Another important trend observed in these approved drugs is that most of these compounds are chiral possessing up to seven stereogenic carbons. Taking into account the FDA requirements for application of chiral drugs, continuous improvements in asymmetric synthesis and characterization of chiral fluorine-containing compounds are of critical importance. In this regard, special attention should be given to a study of the self-disproportionation of enantiomers (SDE), a non-linear behavior of enantiomerically enriched compounds [146–149]. These properties of chiral drugs are an important public safety issue, requiring an additional level of scrutiny in preparation of fluorine-containing drug manufacture [150,151].
Nowadays, fluorine-scans and fluorine-editing are fully embedded activities in medicinal chemistry research, projecting a major impact on contemporary organic chemistry. Therefore, one can expect a continuous increase in the number of fluorinated drugs on the pharmaceutical market in the years to come.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21761132021), the Qing-Lan Project of Jiangsu Province (for Han) and IKERBASQUE, Basque Foundation for Science (for Soloshonok).
| [1] |
J. Fried, E.F. Sabo, J. Am. Chem. Soc. 75 (1953) 2273-2274. DOI:10.1021/ja01105a527 |
| [2] |
J. Fried, E.F. Sabo, J. Am. Chem. Soc. 76 (1954) 1455-1456. |
| [3] |
X.Y. Yang, T. Wu, R.J. Phipps, F.D. Toste, Chem. Rev. 115 (2015) 826-870. DOI:10.1021/cr500277b |
| [4] |
R.G. Syvret, K.M. Butt, T.P. Nguyen, V.L. Bulleck, R.D. Rieth, J. Org. Chem. 67 (2002) 4487-4493. DOI:10.1021/jo020053u |
| [5] |
P. Bravo, S. Capelli, S.V. Meille, et al., Tetrahedron Asymmetry 5 (1994) 2009-2018. DOI:10.1016/S0957-4166(00)86276-X |
| [6] |
X. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 115 (2015) 731-764. DOI:10.1021/cr500193b |
| [7] |
H. Ohkura, D.O. Berbasov, V.A. Soloshonok, Tetrahedron 59 (2003) 1647-1656. DOI:10.1016/S0040-4020(03)00138-8 |
| [8] |
K. Gondo, T. Kitamura, Molecules 17 (2012) 6625-6632. DOI:10.3390/molecules17066625 |
| [9] |
J. Han, A.E. Sorochinsky, T. Ono, V.A. Soloshonok, Curr. Org. Synth. 8 (2011) 281-294. DOI:10.2174/157017911794697277 |
| [10] |
R.G. Syvret, W.J. Casteel, G.S. Lal, J.S. Goudar, J. Fluor. Chem. 125 (2004) 33-35. DOI:10.1016/j.jfluchem.2003.09.002 |
| [11] |
G.V. Röschenthaler, V.P. Kukhar, I.B. Kulik, et al., Tetrahedron Lett. 53 (2012) 539-542. DOI:10.1016/j.tetlet.2011.11.096 |
| [12] |
M.S. Wiehn, E.V. Vinogradova, A. Togni, J. Fluor. Chem. 131 (2010) 951-957. DOI:10.1016/j.jfluchem.2010.06.020 |
| [13] |
M. Shevchuk, Q. Wang, R. Pajkert, et al., Adv. Synth. Catal. 363 (2021) 2912-2968. DOI:10.1002/adsc.202001464 |
| [14] |
E. Merino, C. Nevado, Chem. Soc. Rev. 43 (2014) 6598-6608. DOI:10.1039/C4CS00025K |
| [15] |
J.A. Ma, D. Cahard, J. Fluor. Chem. 128 (2007) 975-996. DOI:10.1016/j.jfluchem.2007.04.026 |
| [16] |
F.L. Qing, X.Y. Liu, J.A. Ma, et al., CCS Chem. 4 (2022) 2518-2549. DOI:10.31635/ccschem.022.202201935 |
| [17] |
S.S. Li, J. Wang, Acta Chim. Sin. 76 (2018) 913-924. DOI:10.6023/A18070306 |
| [18] |
J. Hu, K. Ding, Acta Chim. Sin. 76 (2018) 905-906. DOI:10.6023/A1812E001 |
| [19] |
M. Reichel, K. Karaghiosoff, Angew. Chem. Int. Ed. 59 (2020) 12268-12281. DOI:10.1002/anie.201913175 |
| [20] |
Y. Kraemer, E.N. Bergman, A. Togni, C.R. Pitts, Angew. Chem. Int. Ed. 61 (2022) e202205088. DOI:10.1002/anie.202205088 |
| [21] |
J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432-2506. DOI:10.1021/cr4002879 |
| [22] |
Y. Zhou, J. Wang, Z. Gu, et al., Chem. Rev. 116 (2016) 422-518. DOI:10.1021/acs.chemrev.5b00392 |
| [23] |
Y. Zhu, J. Han, J. Wang, et al., Chem. Rev. 118 (2018) 3887-3964. DOI:10.1021/acs.chemrev.7b00778 |
| [24] |
J. Han, L. Kiss, H. Mei, et al., Chem. Rev. 121 (2021) 4678-4742. DOI:10.1021/acs.chemrev.0c01263 |
| [25] |
H. Mei, J. Han, K.D. Klika, et al., Eur. J. Med. Chem. 186 (2020) 111826. DOI:10.1016/j.ejmech.2019.111826 |
| [26] |
H. Mei, J. Han, S. White, et al., Chem. Eur. J. 26 (2020) 11349-11390. DOI:10.1002/chem.202000617 |
| [27] |
H. Mei, J. Han, S. Fustero, et al., Chem. Eur. J. 25 (2019) 11797-11819. DOI:10.1002/chem.201901840 |
| [28] |
H. Mei, A.M. Remete, Y. Zou, et al., Chin. Chem. Lett. 31 (2020) 2401-2413. DOI:10.1016/j.cclet.2020.03.050 |
| [29] |
S. Purser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320-330. DOI:10.1039/B610213C |
| [30] |
C. Isanbor, D. O’Hagan, J. Fluor. Chem. 127 (2006) 303-319. DOI:10.1016/j.jfluchem.2006.01.011 |
| [31] |
Q. Wang, H. Song, Q. Wang, Chin. Chem. Lett. 33 (2022) 626-642. DOI:10.1016/j.cclet.2021.07.064 |
| [32] |
Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai, N. Shibata, iScience 23 (2020) 101467. DOI:10.1016/j.isci.2020.101467 |
| [33] |
T. Fujiwara, D. O’Hagan, J. Fluor. Chem. 167 (2014) 16-29. DOI:10.1016/j.jfluchem.2014.06.014 |
| [34] |
C. Qin, W. Liu, Y. Nie, et al., Chin. J. Org. Chem. 40 (2020) 2232-2253. DOI:10.6023/cjoc202003051 |
| [35] |
N. Wang, H. Mei, G. Dhawan, et al., Molecule 28 (2023) 3651. DOI:10.3390/molecules28093651 |
| [36] |
Q. Wang, J. Han, A. Sorochinsky, et al., Pharmaceuticals 15 (2022) 999. DOI:10.3390/ph15080999 |
| [37] |
E.P. Gillis, K.J. Eastman, M.D. Hill, D.J. Donnelly, N.A. Meanwell, J. Med. Chem. 58 (2015) 8315-8359. DOI:10.1021/acs.jmedchem.5b00258 |
| [38] |
I. Ojima, J. Fluor. Chem. 198 (2017) 10-23. DOI:10.1016/j.jfluchem.2016.12.016 |
| [39] |
J. Han, A.M. Remete, L.S. Dobson, et al., J. Fluor. Chem. 239 (2020) 109639. DOI:10.1016/j.jfluchem.2020.109639 |
| [40] |
D. O’Hagan, J. Fluor. Chem. 131 (2010) 1071-1081. DOI:10.1016/j.jfluchem.2010.03.003 |
| [41] |
Y. Yu, A. Liu, G. Dhawan, et al., Chin. Chem. Lett. 32 (2021) 3342-3354. DOI:10.1016/j.cclet.2021.05.042 |
| [42] |
J. He, Z. Li, G. Dhawan, et al., Chin. Chem. Lett. 34 (2023) 107578. DOI:10.1016/j.cclet.2022.06.001 |
| [43] |
B. Aslan, G. Kismali, L.R. Iles, et al., Blood Cancer J. 12 (2022) 80. |
| [44] |
E.B. Gomez, K. Ebata, H.S. Randeria, et al., Blood 142 (2023) 62-72. DOI:10.1182/blood-2023-183029 |
| [45] |
P. Thompson, C. Tam, Blood 141 (2023) 3137-3142. |
| [46] |
D. Telaraja, Y.L. Kasamon, J.S. Collazo, et al., Clin. Cancer Res. 30 (2024) 17-22. DOI:10.1158/1078-0432.CCR-23-1272 |
| [47] |
A.R. Mato, N.V. Shah, W. Jurczak, et al., Lancet 397 (2021) 892-901. DOI:10.1016/S0140-6736(21)00224-5 |
| [48] |
S.J. Keam, Drugs 83 (2023) 547-553. DOI:10.1007/s40265-023-01860-1 |
| [49] |
D. Arguelles, J. Alonso, C.T. Eary, et al., Patent, WO 2022056100 A1, 2022.
|
| [50] |
A. Cuadrado, G. Manda, A. Hassan, et al., Pharmacol. Rev. 70 (2018) 348-383. DOI:10.1124/pr.117.014753 |
| [51] |
S. Saha, B. Buttari, E. Panieri, E. Profumo, L. Saso, Molecules 25 (2020) 5474. DOI:10.3390/molecules25225474 |
| [52] |
R. Borella, L. Forti, L. Gibellini, et al., Molecules 24 (2019) 4097. DOI:10.3390/molecules24224097 |
| [53] |
T. Shekh-Ahmad, R. Eckel, S.D. Naidu, et al., Brain 141 (2018) 1390-1403. DOI:10.1093/brain/awy071 |
| [54] |
H. Sun, J. Zhu, H. Lin, K. Gu, F. Feng, Expert Opin. Ther. Pat. 27 (2017) 763-785. DOI:10.1080/13543776.2017.1325464 |
| [55] |
A. Lee, Drugs 83 (2023) 725-729. DOI:10.1007/s40265-023-01874-9 |
| [56] |
V. Profeta, K. McIntyre, M. Wells, C. Park, D.R. Lynch, Expert Opin. Investig. 32 (2023) 5-16. DOI:10.1080/13543784.2023.2173063 |
| [57] |
E. Anderson, A. Decker, X. Liu, Patent, WO 2013163344 A1, 2013.
|
| [58] |
M.B. Sporn, K.T. Liby, M.M. Yore, et al., J. Nat. Prod. 74 (2011) 537-545. DOI:10.1021/np100826q |
| [59] |
K. Hoegenauer, N. Soldermann, F. Zécri, et al., ACS Med. Chem. Lett. 8 (2017) 975-980. DOI:10.1021/acsmedchemlett.7b00293 |
| [60] |
C. Bauer, T. Luu, F. Eggimann, et al., Helv. Chim. Acta 101 (2018) e1800044. DOI:10.1002/hlca.201800044 |
| [61] |
Z. Wu, X. Li, X. Chen, et al., Front. Pharmacol. 13 (2022) 1021714. |
| [62] |
J. Zhang, H. Jiang, S. Lin, et al., J. Med. Chem. 65 (2022) 8011-8028. DOI:10.1021/acs.jmedchem.2c00530 |
| [63] |
S. Duggan, Z.T. Al-Salama, Drugs 83 (2023) 943-948. DOI:10.1007/s40265-023-01895-4 |
| [64] |
P.A. Fernandes Gomes Dos Santos, K. Högenauer, G. Holling-Worth, et al., Patent, WO 2013001445 A1, 2013.
|
| [65] |
N.G. Cooke, P.A. Fernandes Gomes Dos Santos, P. Furet, et al., Patent, WO 2013088404 A1, 2013.
|
| [66] |
M. Beier, D. Willen, S. Krösser, T. Schlüter, Patent, WO 2020058504 A1, 2020.
|
| [67] |
T. Keller, D. Röthlein, J. Schmitt, et al., Patent, WO 2014154531 A1, 2014.
|
| [68] |
P. Ivanov Bichovski, F. Löscher, S. Nicoletti, Patent, WO 2018228975 A1, 2018.
|
| [69] |
A. Ballesteros-Sánchez, C. De-Hita-Cantalejo, M.C. Sánchez-González, et al., Ocul. Surf. 30 (2023) 254-262. DOI:10.1016/j.jtos.2023.10.001 |
| [70] |
Y.K. Kim, B. Gonther, H. Meinert, Eur. J. Ophthalmol. 15 (2005) 627-637. DOI:10.1177/112067210501500514 |
| [71] |
T.D. Le, R.A. Arlauskas, J.G. Weers, J. Fluor. Chem. 78 (1996) 153-156. |
| [72] |
S. Barata-Vallejo, A. Postigo, J. Org. Chem. 75 (2010) 6141-6148. DOI:10.1021/jo100901z |
| [73] |
H. Depypere, D. Timmerman, G. Donders, et al., J. Clin. Endocr. Metab. 104 (2019) 5893-5905. DOI:10.1210/jc.2019-00677 |
| [74] |
H.R. Hoveyda, G.L. Fraser, M.O. Roy, et al., J. Med. Chem. 58 (2015) 3060-3082. DOI:10.1021/jm5017413 |
| [75] |
H.R. Hoveyda, G.L. Fraser, G. Dutheuil, et al., ACS Med. Chem. Lett. 6 (2015) 736-740. DOI:10.1021/acsmedchemlett.5b00117 |
| [76] |
A. Lee, Drugs 83 (2023) 1137-1141. DOI:10.1007/s40265-023-01917-1 |
| [77] |
A.N. Comninos, W.S. Dhillo, Cell 186 (2023) 3332-3332.e1. DOI:10.1016/j.cell.2023.07.011 |
| [78] |
H. Hoveyda, G. Dutheuil, G. Fraser, et al., Patent, WO 201305424 A1, 2013.
|
| [79] |
J. Gerhart, D.S. Cox, R.S.P. Singh, et al., Clin. Pharmacokinet. 63 (2024) 27-42. DOI:10.1007/s40262-023-01339-y |
| [80] |
K. Anand, J. Ziebuhr, P. Wadhwani, J.R. Mesters, R. Hilgenfeld, Science 300 (2003) 1763-1767. DOI:10.1126/science.1085658 |
| [81] |
J. Hammond, H. Leister-Tebbe, A. Gardner, et al., N. Engl. J. Med. 386 (2022) 1397-1408. DOI:10.1056/NEJMoa2118542 |
| [82] |
D.R. Owen, C.M.N. Allerton, A.L.S. Anderson, et al., Science 374 (2021) 1586-1593. DOI:10.1126/science.abl4784 |
| [83] |
I.F. Sevrioukova, T.L. Poulos, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 18422-18427. DOI:10.1073/pnas.1010693107 |
| [84] |
C. Shekhar, R. Nasam, S.R. Paipuri, et al., Tetrahedron Chem. 4 (2022) 100033. DOI:10.1016/j.tchem.2022.100033 |
| [85] |
Y.R. Alugubelli, Z.Z. Geng, K.S. Yang, et al., Eur. J. Med. Chem. 240 (2022) 114596. DOI:10.1016/j.ejmech.2022.114596 |
| [86] |
D.R. Owen, M.Y. Pettersson, M.R. Reese, et al., Patent, US 11351149 B2, 2022.
|
| [87] |
D.R. Owen, M.Y. Pettersson, M.R. Reese, et al., Patent, WO 2021250648 A1, 2021.
|
| [88] |
B.A. Cotrim, J.C. Barros, Aust. J. Chem. 75 (2022) 487-491. DOI:10.1071/CH22104 |
| [89] |
W.L. Shoop, E.J. Hartline, B.R. Gould, et al., Vet. Parasitol. 201 (2014) 179-189. DOI:10.1016/j.vetpar.2014.02.020 |
| [90] |
M.P. Curtis, V. Vaillancourt, R.M. Goodwin, et al., Bioorg. Med. Chem. Lett. 26 (2016) 1831-1835. DOI:10.1016/j.bmcl.2016.02.027 |
| [91] |
L. Rufener, V. Danelli, D. Bertrand, et al., Parasites Vectors 10 (2017) 530. DOI:10.1186/s13071-017-2470-4 |
| [92] |
Y.Y. Gao, M.A. Di Pascuale, W. Li, et al., Investig. Ophthalmol. Vis. Sci. 46 (2005) 3089-3094. DOI:10.1167/iovs.05-0275 |
| [93] |
W. Trattler, P. Karpecki, Y. Rapoport, et al., Clin. Ophthalmol. 16 (2022) 1153-1164. DOI:10.2147/OPTH.S354692 |
| [94] |
L. O’Dell, D.S. Dierker, D.K. Devries, et al., Clin. Ophthalmol. 16 (2022) 2979-2987. DOI:10.2147/OPTH.S374530 |
| [95] |
E. Yeu, D.L. Wirta, P. Karpecki, et al., Cornea 42 (2022) 435-443. |
| [96] |
C.E. Toutain, W. Seewald, M. Jung, Parasites Vectors 11 (2018) 412. DOI:10.1186/s13071-018-2966-6 |
| [97] |
C.E. Toutain, W. Seewald, M. Jung, Parasites Vectors 10 (2017) 522. DOI:10.1186/s13071-017-2475-z |
| [98] |
L. Rufener, V. Danelli, D. Bertrand, H. Sager, Parasites Vectors 10 (2017) 530. DOI:10.1186/s13071-017-2470-4 |
| [99] |
N. Lamassiaude, B. Toubate, C. Neveu, et al., PLoS Pathog. 17 (2021) e1008863. DOI:10.1371/journal.ppat.1008863 |
| [100] |
S. Nanchen, Patent, WO2014090918 A1, 2014.
|
| [101] |
S. Dhillon, Drugs 24 (2024), doi: 10.1007/s40265-023-01990-6.
|
| [102] |
A. Drilon, S.H.I. Ou, B.C. Cho, et al., Cancer Discov. 8 (2018) 1227-1236. DOI:10.1158/2159-8290.CD-18-0484 |
| [103] |
J.J. Cui, Y. Li, E.W. Rogers, Patent, WO2017004342 A1, 2017.
|
| [104] |
J. Li, D. Zhang, J. Feng, et al., Patent, WO2019201282 A1, 2019.
|
| [105] |
J.A. Ellman, Pure Appl. Chem. 75 (2023) 39-46. |
| [106] |
M.T. Robak, M.A. Herbage, J.A. Ellman, Chem. Rev. 110 (2010) 3600-3740. DOI:10.1021/cr900382t |
| [107] |
C. Xie, L. Wu, H. Mei, Org. Biomol. Chem. 12 (2014) 7836-7843. DOI:10.1039/C4OB01575D |
| [108] |
J.L. Moore, S.M. Taylor, V.A. Soloshonok, Arkivoc 6 (2005) 287-292. |
| [109] |
S. Chen, J. Xu, Tetrahedron Lett. 32 (1991) 6711-6714. DOI:10.1016/S0040-4039(00)93583-4 |
| [110] |
L. Xu, G. Wang, N. Rong, et al., Org. Process Res. Dev. (2023). DOI:10.1021/acs.oprd.3c00152 |
| [111] |
A.E. Sorochinsky, J.L. Aceña, V.A. Soloshonok, Synthesis 45 (2013) 141-152. |
| [112] |
T. Nakamura, K. Tateishi, S. Tsukagoshi, et al., Tetrahedron 68 (2012) 4013-4017. DOI:10.1016/j.tet.2012.03.054 |
| [113] |
Y. Suzuki, J. Han, O. Kitagawa, et al., RSC Adv. 5 (2015) 2988-2993. DOI:10.1039/C4RA13928C |
| [114] |
M.A. Brodney, D.D. Auperin, S.L. Becker, et al., Bioorg. Med. Chem. Lett. 21 (2011) 2637-2640. DOI:10.1016/j.bmcl.2010.12.118 |
| [115] |
T. Takahashi, J.R. Prensner, C.D. Robson, K.A. Janeway, B.J. Weigel, Pediatr. Blood Cancer 67 (2020) e28636. DOI:10.1002/pbc.28636 |
| [116] |
S. Lonial, S. Grosicki, M. Hus, et al., J. Clin. Oncol. 40 (2022) 8019. DOI:10.1200/JCO.2022.40.16_suppl.8019 |
| [117] |
M. Gounder, R. Ratan, T. Alcindor, et al., N. Engl. J. Med. 388 (2023) 898-912. DOI:10.1056/NEJMoa2210140 |
| [118] |
K. Patterson, M. Hatcher, Patent, WO 2023034917 A1, 2023.
|
| [119] |
E. Jonasch, F. Donskov, O. Iliopoulos, et al., N. Engl. J. Med. 385 (2021) 2036-2046. DOI:10.1056/NEJMoa2103425 |
| [120] |
T.K. Choueiri, T.M. Bauer, K.P. Papadopoulos, et al., Nat. Med. 27 (2021) 802-805. DOI:10.1038/s41591-021-01324-7 |
| [121] |
V. Visweswaran, K. Pavithran, Curr. Drug Res. Rev. 14 (2022) 88-95. DOI:10.2174/2589977514666220401094724 |
| [122] |
R. Xu, K. Wang, J.P. Rizzi, et al., J. Med. Chem. 62 (2019) 6876-6893. DOI:10.1021/acs.jmedchem.9b00719 |
| [123] |
E.D. Deeks, Drugs 81 (2021) 1921-1927. DOI:10.1007/s40265-021-01606-x |
| [124] |
K.B. Hamal, C.I. Pavlich, G.J. Carlson, et al., Tetrahedron Lett. 128 (2023) 154691. DOI:10.1016/j.tetlet.2023.154691 |
| [125] |
J.P. Bégué, D. Bonnet-Delpon, J. Fluor. Chem. 127 (2006) 992-1012. DOI:10.1016/j.jfluchem.2006.05.006 |
| [126] |
S. Akyuz, A.E. Ozel, K. Balci, et al., J. Mol. Struct. 993 (2011) 319-323. DOI:10.1016/j.molstruc.2011.01.041 |
| [127] |
M. Boberg, A.C. Jonson, H. Leek, R. Jansson-Löfmark, M. Ashton, ACS Omega 5 (2020) 23885-23891. DOI:10.1021/acsomega.0c03121 |
| [128] |
B.W. Metcalf, P. Bey, C. Danzin, et al., J. Am. Chem. Soc. 100 (1978) 2551-2553. DOI:10.1021/ja00476a050 |
| [129] |
C. Burri, R. Brun, Parasitol. Res. 90 (2003) S49-S52. DOI:10.1007/s00436-002-0766-5 |
| [130] |
B. Bouteille, O. Oukem, S. Bisser, M. Dumas, Fundam. Clin. Pharmacol. 17 (2003) 171-181. DOI:10.1046/j.1472-8206.2003.00167.x |
| [131] |
M.P. Barrett, I.M. Vincent, R.J.S. Burchmore, A.J.N. Kazibwe, E. Matovu, Future Microbiol. 6 (2011) 1037-1047. DOI:10.2217/fmb.11.88 |
| [132] |
J. Zhu, B.A. Price, J. Walker, S.X. Zhao, Tetrahedron Lett. 46 (2005) 2795-2797. DOI:10.1016/j.tetlet.2005.02.146 |
| [133] |
K.P. Garnock-Jones, Drugs 75 (2015) 1645-1656. DOI:10.1007/s40265-015-0463-1 |
| [134] |
M. Sanford, Drugs 70 (2010) 1615-1627. DOI:10.2165/11205930-000000000-00000 |
| [135] |
A. Hatzelmann, E.J. Morcillo, G. Lungarella, et al., Pulm. Pharmacol. Ther. 23 (2010) 235-256. DOI:10.1016/j.pupt.2010.03.011 |
| [136] |
M.A. Giembycz, S.K. Field, Drug Des. Dev. Ther. 4 (2010) 147-158. |
| [137] |
K.F. Rabe, Br. J. Pharmacol. 163 (2011) 53-67. DOI:10.1111/j.1476-5381.2011.01218.x |
| [138] |
L.A. Sorbera, P.A. Leeson, J. Castaner, Drug Future 25 (2000) 1261-1264. DOI:10.1358/dof.2000.025.12.604045 |
| [139] |
G.L. Card, B.P. England, Y. Suzuki, et al., Structure 12 (2004) 2233-2247. DOI:10.1016/j.str.2004.10.004 |
| [140] |
A. Hermann, Patent, WO9501338 A1, 1993.
|
| [141] |
Y.J. Chen, S. Pikul, S.C. Kuo, G.D. Chu, Patent, WO 2013131484 A1, 2013.
|
| [142] |
Y.J. Chen, S. Pikul, Patent, WO 2013131255 A1, 2013.
|
| [143] |
P. Vijaykar, D. Kokane, S. Gharat, et al., Patent, US 20140275551 A1, 2014.
|
| [144] |
Y. Lin, P. Huang, S. Liu, et al., Res. Chem. Intermed. 39 (2013) 2107-2113. DOI:10.1007/s11164-012-0742-3 |
| [145] |
G. Amari, E. Armani, E. Ghidini, Patent, US 20080015226 A1, 2008.
|
| [146] |
J. Han, O. Kitagawa, A. Wzorek, K.D. Klika, V.A. Soloshonok, Chem. Sci. 9 (2018) 1718-1739. DOI:10.1039/C7SC05138G |
| [147] |
J. Han, D.J. Nelson, A.E. Sorochinsky, V.A. Soloshonok, Curr. Org. Synth. 8 (2011) 310-317. DOI:10.2174/157017911794697303 |
| [148] |
A.E. Sorochinsky, T. Katagiri, T. Ono, et al., Chirality 25 (2013) 365-368. DOI:10.1002/chir.22180 |
| [149] |
H. Ueki, M. Yasumoto, V.A. Soloshonok, Tetrahedron Asymmetry 21 (2010) 1396-1400. DOI:10.1016/j.tetasy.2010.04.040 |
| [150] |
J. Han, A. Wzorek, K.D. Klika, V.A. Soloshonok, Molecules 26 (2021) 2757. DOI:10.3390/molecules26092757 |
| [151] |
J. Han, R. Dembinski, V.A. Soloshonok, K.D. Klika, Molecules 26 (2021) 3994. DOI:10.3390/molecules26133994 |