 2024, Vol. 35
2024, Vol. 35 


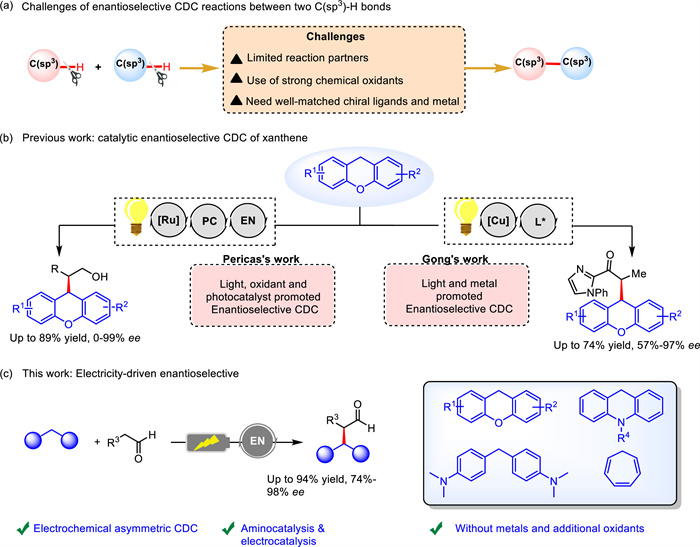
As an efficient, safe and sustainable synthetic strategy, electrocatalytic coupling reaction has received unprecedented attention in modern practical organic synthesis [1–11]. Carbon-carbon bond coupling reaction, especially those involving two C(sp3) atoms, is one of the most commonly used reactions in the synthesis of drug molecules and material molecules [12–15]. Recently, using functionalized carbon sources as substrates, a series of excellent electrocatalytic carbon-carbon bond coupling reactions have been reported [16–19]. For instance, Baran's group used redox-active esters and aromatic halide to realize decarboxylative cross-coupling [58]. In view of the abundance of C(sp3)–H bonds in organic compounds, direct cross-dehydrogenative coupling (CDC) of two C(sp3)–H bonds has attracted great attention since it avoids prior functionalization of the substrates, thereby providing atom and step economy for C–C bond formation [20–25]. However, there are still several challenges associated with this strategy (Scheme 1a). One of the critical holdback is the typical requirement of harsh conditions including the use of strong chemical oxidants such as 2,3-dicyano-5,6-dichlorobenzoquinone (DDQ), tert-butyl hydroperoxide (TBHP) or ammonium persulfate to activate the inert C(sp3)–H bonds [26–32]. Moreover, their catalytic asymmetric variants require the design of well-matched chiral ligands, metal catalysts and substrates. As a result, a handful of methods for catalytic enantioselective CDC have been reported (Scheme 1b) [24,59]. Despite asymmetric CDC of two C(sp3)–H bonds involving electrocatalysis with metal catalysts has been reported [33–40], development of this reaction with metal-free catalysts and even without additional oxidants is fascinating and has rarely been explored [41–42,52–57].

|
Download:
|
| Scheme 1. Asymmetric cross-dehydrogenative coupling reaction of two C(sp3)-H bonds. | |
Recently, organocatalysis as stable organic molecules has emerged as a powerful source of enantioselective transformations and has attracted great interest from chemists [43–45]. Particularly, aminocatalysis has been successfully employed to enable highly stereoselective C−H functionalizations by synergistic coupling with established C−H activation/functionalization cycles [46–48]. However, there are still many problems and challenges in this field: the catalytic efficiency is relatively low, often requires a higher amount of catalyst, increasing the cost of the reaction and the difficulty of subsequent purification. Compared with metal catalysis, its catalytic system and reaction types are limited, which limits the application of asymmetric organic catalysis [49–51]. Chiral amine catalysis via an enamine intermediate has been established in electrochemical processes [52–57], which encouraged us to further explore new asymmetric CDC of two C(sp3)–H bonds by combining electrocatalysis and organocatalysis.
Herein, we present a more extensive asymmetric synthesis using amine-based chiral catalysts under electrochemical conditions, which can directly achieve CDC of two C(sp3)-H bonds without the involvement of metals and oxidants (Scheme 1c). A series of alkyl aldehydes and various substrates containing C(sp3)-H bonds, including xanthenes, acridines, cycloheptatrienes and even diarylmethane have been shown to undergo asymmetric CDC to afford a series of carbon-carbon bond coupling products with up to 94% yield and 98% ee.
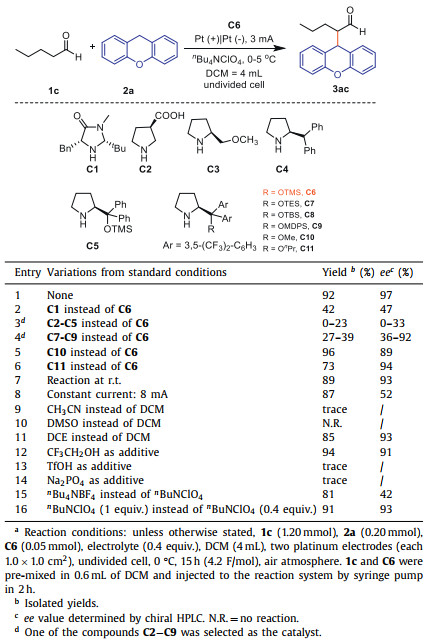
To investigate whether amine-based chiral catalysts can be used to catalyze the enantioselective CDC of aldehydes under electrochemical conditions, we chosed n-pentanal with xanthene as the template substrate to explore the conditions (Table 1, for more details, see Table S1 in Supporting information). Firstly, we tested imidazolidinone as chiral catalyst (C1), which got product 3ac in 42% yield and 47% ee (Table 1, entry 2). Then we tried various amine-based chiral catalysts, to our delight, 3ac could be obtained in 92% yield when C6 was used as catalyst (Table 1, entry 1). In contrast, catalysts C2-C5 could only obtain the product 3ac in 0–23% yields and 0–33% ee (Table 1, entry 3, for more details, see Table S1, entries 2–5). Subsequently, we examined the effect of different l-proline derivatives on the yields and ee values when the substituent was changed. Catalysts C7-C9 afforded the product in 27%−39% yields and 36%−92% ee (Table 1, entry 4, for more details, see Table S1, entries 7–9). Specifically, when the substituent group was changed to a methyl group with small steric hindrance (C10), the target product 3ac could be obtained in 96% yield, but the ee value slightly reduced (Table 1, entry 5). Subsequently, we changed the substituent group to n-propyl (C11), the ee value reached to 94%, while the yield decreased to 73% (Table 1, entry 6). Reaction at room temperature (Table 1, entry 7) led to a decrease in yield and ee value. When the reaction was carried out at a constant current of 8 mA, the ee value decreased sharply (Table 1, entry 8). When changing to other solvents, the reaction was not as effective as DCM (Table 1, entries 9–11). Next, we attempted to promote the C—C bond formation by adding proton additives in order to increase the generation of the active intermediate enamine.
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
Unfortunately, when trifluoroethanol was added, the yield was slightly increased but the ee value decreased to 91% (Table 1, entry 12). Other additives, such as TfOH, led to a dramatic decrease in yield (Table 1, entry 13). Then we tried adding a base to the reaction system but only found little product (Table 1, entry 14). We also studied different electrolytes and studied the amount of electrolyte, none of which were more effective than standard conditions (Table 1, entries 15 and 16).
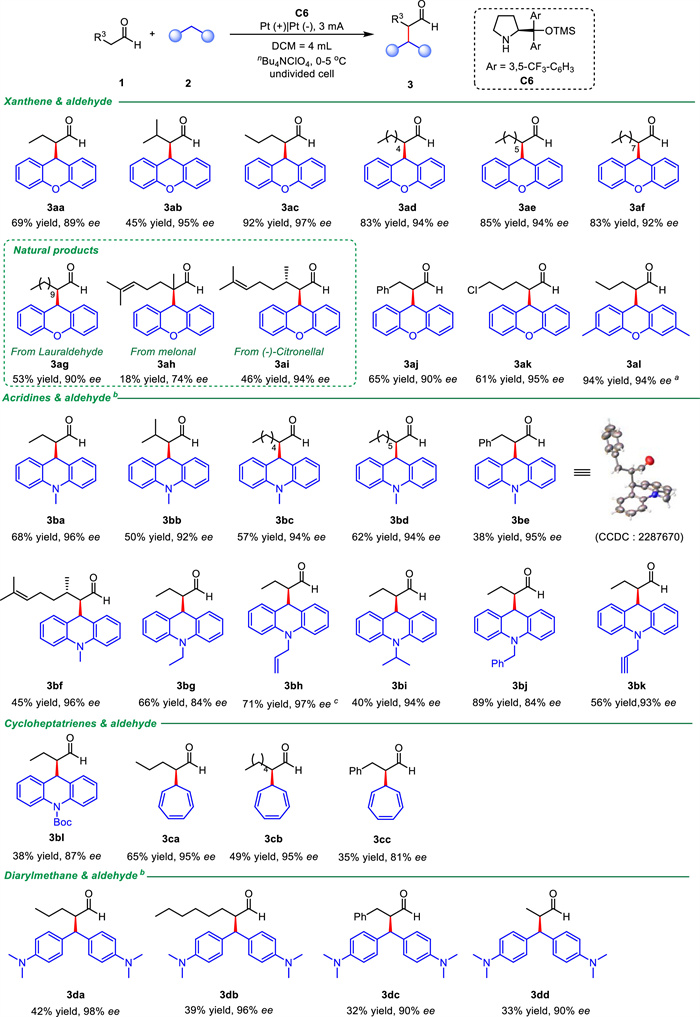
Under optimal conditions, the substrate range of aldehydes and C(sp3)-H were investigated separately. Firstly, the substrate range of aldehydes was extended (Scheme 2). From short-chain aliphatic aldehydes to long-chain aliphatic aldehydes were obtained to the target products (3aa-3ag) in high yields and ee values. Surprisingly, the natural product 2,6-dimethyl-5-heptenal with a large steric hindrance also gave the product 3ah in 18% yield and 74% ee value. In contrast, the natural product (-)-citronellal was able to reach the target product 3ai in 46% yield and 94% ee value. In addition, phenylpropanal and chloropentanal with halogen atoms were also compatible with this electrochemically catalyzed reaction system, yielding good yields and high ee values (3aj and 3ak). Further more, xanthene derivatives can also have high compatibility with aldehyde, obtaining 94% yield and 94% ee value (3al).

|
Download:
|
| Scheme 2. Substrate scope of aldehydes and C(sp3)-H: Reaction conditions: unless otherwise stated, 1c (1.20 mmol), 2a (0.20 mmol), C6 (0.05 mmol), electrolyte (0.4 equiv.), DCM (4 mL), two platinum electrodes (each 1.0 × 1.0 cm2), undivided cell, 0–5 ℃, 15 h (4.2 – 1.1 F/mol), air atmosphere. 1c and C6 were pre-mixed in 0.6 mL of DCM and injected to the reaction system by syringe pump in 2 h. Isolated yields. The ee values were determined by chiral HPLC analysis. a Reacts for 9 h. b Constant current: 1 mA; reacts for 12 h; TFA = 20 mol%. c Reacts for 10 h. | |
{kind=link}
Secondly, we examined the range of substrates with different C(sp3)-H. Methylacridine could be compatible with different aliphatic and aromatic aldehydes (3ba-3be), where we determined the absolute configuration of 3be to be the R configuration by single-crystal X-ray diffraction (Fig. S2 in Supporting information for details). Methylacridine can also reacted with (-)-Citronellal gave a yield of 45%, 95% ee (3bf). Electron-donating (3bg-3bk) or electron-withdrawing (3bl) substituents at the N position of the acridine were studied. The yield of 3bl is reduced while all products provided good ee values. In addition, cycloheptatriene and aliphatic aldehydes could also reach the target products 3ca and 3cb with high ee values. However, cycloheptatriene with phenylpropanal could only reach 3cc with 35% yield and 81% ee value. To our surprise, the ring-opened diarylmethane was also able to give the product 3da-3dd under these reaction conditions in 42%−32% yield and 98%−90% ee value.
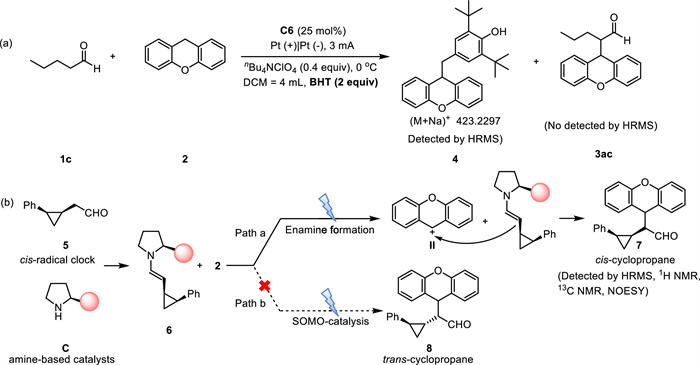
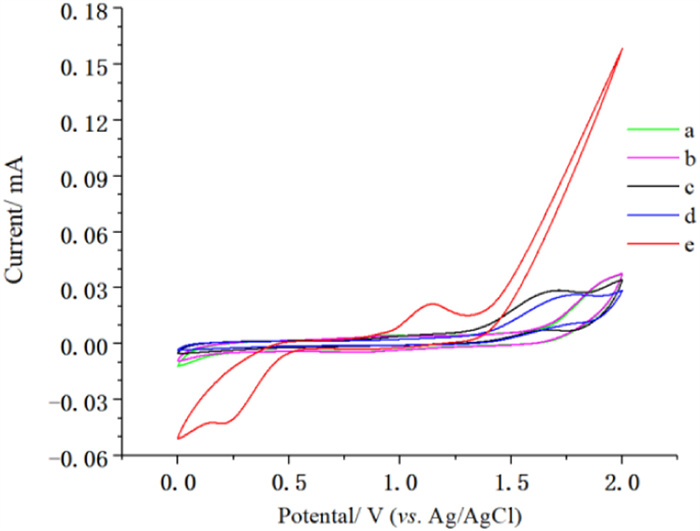
Subsequently, we investigated the reaction mechanism. Adding 2 equiv. BHT to the optimal reaction system resulted in no detection of 3ac, but products 4 were detected under BHT by using HRMS (Scheme 3a). Based on the above control experiments, we performed cyclic voltammetry experiments (Fig. 1). As shown in Fig. 1, the oxidation peak of 2 was observed at 1.15 V vs. Ag/AgCl (Fig. 1e, red line) which was lower than others. The obtained results from CV experiment supported that xanthene 2 was easier to be oxidized comparing with aldehyde (Fig. 1b, pink line) and enamine intermediates (Fig. 1c, black line).

|
Download:
|
| Scheme 3. Control experiments. | |
{kind=link}

|
Download:
|
| Fig. 1. Cyclic voltammograms. The cyclic voltammograms were recorded in an electrolyte solution of n Bu4NClO4 (0.1 mol/L) in DCM (5 mL) using a glassy carbon disk working electrode (diameter, 3 mm), a Pt wire auxiliary electrode and an Ag/AgCl reference electrode. The scan rate was 100 mV/s. (a) background, (b) 1c (10 mmol/L); (c) 1c (10 mmol/L) + C6 (2.5 mmol/L); (d) C6 (2.5 mmol/L); (e) 2 (10 mmol/L). | |
{kind=link}
Then we implemented a suitable radical clock experiment which allowed us to demonstrate that only xanthene was oxidized under electrochemical conditions [48,60]. When the cis-cyclopropyl-substituted aldehyde 5 was exposed to the alkylation protocol in the presence of xanthene 2, the cis-substituted cyclopropane product 7 was formed exclusively (Scheme 3b, for more details see 6.2 Radical clock experiment in Supporting information). Trans-substituted cyclopropane product 8 was not generated in this experiment. This result suggested that the reaction occurred through an enamine addition mechanism (Path a), enamine was not oxidized at the anode to form a radical. Therefore, we considered that xanthene 2 underwent anode oxidation to generate a cation intermediate Ⅱ which reacted with enamine through addition reaction.
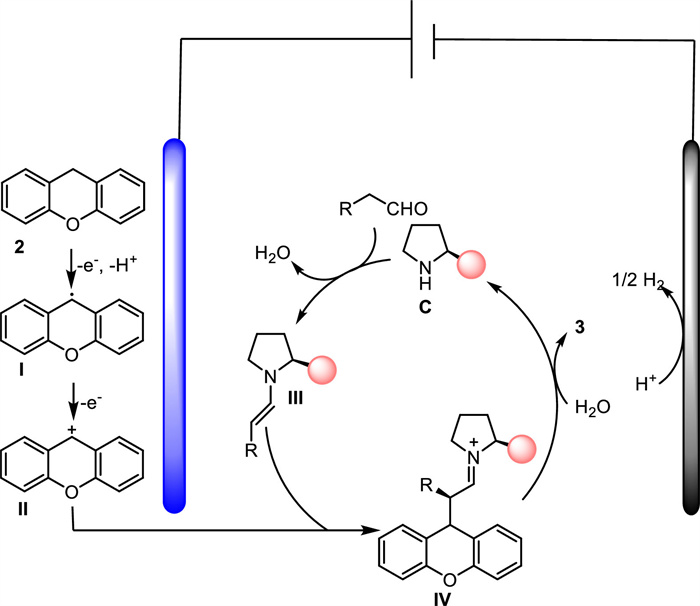
Based on the above experiments and literature precedent, a catalytic cycle for this asymmetric electrochemical process was proposed (Scheme 4). Initially, xanthene 2 underwent anodic oxidation to generate radical species Ⅰ, which would be further oxidized into a cation intermediate Ⅱ by losing another electron. Meanwhile, condensation of C with aldehyde formed enamine Ⅲ, and nucleophilic attack of Ⅱ by Ⅲ afforded iminium Ⅳ. Ⅳ could be hydrolyzed to release coupling product 3 and regenerated organocatalyst C. Finally, protons were reduced to hydrogen at the cathode.

|
Download:
|
| Scheme 4. Proposed reaction mechanism. | |
{kind=link}
In summary, we have established an efficient catalytic asymmetric electrochemical approach for the CDC reaction of aldehydes with different C(sp3)-H bonds by anodic oxidation and organocatalysis which has high enantioselectivity and good yield. Notably, the electrocatalytic asymmetric CDC reactions of acridines and diarylmethane have not been reported before. This protocol operates in the absence of stoichiometric additives, including metals and oxidants, which contributes to its broad substrate range. Traditional CDC often require transition metal catalysis, whereas harsh conditions, such as stoichiometric oxidants or elevated temperatures, may deliver inevitable wastes. Therefore, we proposed a pathway which was put forward in the absence of external oxidants with hydrogen evolution to substantially improve atom economy.
However, the method is unsuitable for broader range of ring-opening substrates. Reactions of diphenylmethane or bis(4-methoxyphenyl)methane failed to generate the desired products. In the future, electricity-driven enantioselective reactions can be further developed to broaden the substrate range of diarylmethane derivatives.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. 22161008, 22061003), Guangxi Science and Technology Base and Talent Project (High Level Innovative Talents and Team Training) (Guike No. AD23026094), Guangxi Natural Science Foundation of China (No. 2021GXNSFFA220005) for financial support.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109445.
| [1] |
Y. Yuan, A. Lei, Acc. Chem. Res. 52 (2019) 3309-3324. DOI:10.1021/acs.accounts.9b00512 |
| [2] |
Z. Yang, W. Shi, H. Alhumade, H. Yi, A. Lei, Nat. Synth. 2 (2023) 217-230. DOI:10.1038/s44160-022-00221-2 |
| [3] |
J.L. Röckl, D. Pollok, R. Franke, S.R. Waldvogel, Acc. Chem. Res. 53 (2020) 45-61. DOI:10.1021/acs.accounts.9b00511 |
| [4] |
W.B. He, S.J. Zhao, J.Y. Chen, et al., Chin. Chem. Lett. 34 (2023) 107640. DOI:10.1016/j.cclet.2022.06.063 |
| [5] |
Y.H. Lu, S.Y. Mu, J. Jiang, et al., ChemSusChem 16 (2023) e202300523. DOI:10.1002/cssc.202300523 |
| [6] |
H.T. Tang, Y.Z. Pan, Y.M. Pan, Green Chem. 25 (2023) 8313-8327. DOI:10.1039/d3gc02106h |
| [7] |
Y.H. Lu, Z.T. Zhang, H.Y. Wu, et al., Chin. Chem. Lett. 34 (2023) 108036. DOI:10.1016/j.cclet.2022.108036 |
| [8] |
X.Q. Zhou, H.T. Tang, F.H. Cui, et al., Green Chem. 25 (2023) 5024-5029. DOI:10.1039/d3gc01288c |
| [9] |
J.Y. Chen, C.T. Zhong, Q.W. Gui, et al., Chin. Chem. Lett. 32 (2021) 475-479. DOI:10.1016/j.cclet.2020.09.034 |
| [10] |
Z.L. Wu, J.Y. Chen, X.Z. Tian, et al., Chin. Chem. Lett. 33 (2022) 1501-1504. DOI:10.1016/j.cclet.2021.08.071 |
| [11] |
Y. Wu, J.Y. Chen, H.R. Liao, et al., Green Synth. Catal. 2 (2021) 233-236. DOI:10.1016/j.gresc.2021.03.006 |
| [12] |
I. Kerschgens, A.R. Rovira, R. Sarpong, J. Am. Chem. Soc. 140 (2018) 9810-9813. DOI:10.1021/jacs.8b05832 |
| [13] |
X.Y. Dong, Y.F. Zhang, C.L. Ma, et al., Nat. Chem. 11 (2019) 1158-1166. DOI:10.1038/s41557-019-0346-2 |
| [14] |
Y. Gao, C. Yang, S. Bai, et al., Chem 6 (2020) 675-688. DOI:10.1016/j.chempr.2019.12.010 |
| [15] |
Y.H. Lu, S.Y. Mu, H.X. Li, et al., Green Chem. 25 (2023) 5539. DOI:10.1039/d2gc04906f |
| [16] |
W. Zhang, L. Lu, W. Zhang, et al., Nature 604 (2022) 292-297. DOI:10.1038/s41586-022-04540-4 |
| [17] |
B. Zhang, Y. Gao, Y. Hioki, et al., Nature 606 (2022) 313-318. DOI:10.1038/s41586-022-04691-4 |
| [18] |
M.Y.S. Ibrahim, G.R. Cumming, D.V.R. Gonzalez, et al., J. Am. Chem. Soc. 145 (2023) 17023-17028. DOI:10.1021/jacs.3c07313 |
| [19] |
S. Mu, H. Lia, Z. Wu, et al., Chin. J. Org. Chem. 42 (2022) 4292. DOI:10.6023/cjoc202211002 |
| [20] |
T. Tian, Z. Li, C.J. Li, Green Chem. 23 (2021) 6789-6862. DOI:10.1039/d1gc01871j |
| [21] |
K. Matcha, A.P. Antonchick, Angew. Chem. Int. Ed. 52 (2013) 2082-2086. DOI:10.1002/anie.201208851 |
| [22] |
Z.J. Wu, H.C. Xu, Angew. Chem. Int. Ed. 56 (2017) 4734-4738. DOI:10.1002/anie.201701329 |
| [23] |
S.A. Girard, T. Knauber, C.J. Li, Angew. Chem. Int. Ed. 53 (2014) 74-100. DOI:10.1002/anie.201304268 |
| [24] |
E. Larionov, M.M. Mastandrea, M.A. Pericàs, ACS Catal. 7 (2017) 7008-7013. DOI:10.1021/acscatal.7b02659 |
| [25] |
A.K. Bagdi, M. Rahman, D. Bhattacherjee, et al., Green Chem. 22 (2020) 6632-6681. DOI:10.1039/d0gc02437f |
| [26] |
J. Blom, G.J. Reyes-Rodríguez, H.N. Tobiesen, et al., Angew. Chem. Int. Ed. 58 (2019) 17856-17862. DOI:10.1002/anie.201911793 |
| [27] |
B. Zhang, S.K. Xiang, L.H. Zhang, Y. Cui, N. Jiao, Org. Lett. 13 (2011) 5212-5215. DOI:10.1021/ol202090a |
| [28] |
Q. Chen, X. Wang, G. Yu, C. Wen, Y. Huo, Org. Chem. Front. 5 (2018) 2652-2656. DOI:10.1039/c8qo00740c |
| [29] |
W. Cao, X. Liu, R. Peng, et al., Chem. Commun. 49 (2013) 3470-3472. DOI:10.1039/c3cc41315b |
| [30] |
Y. Wei, J. Tang, X. Cong, X. Zeng, Green Chem. 15 (2013) 3165-3169. DOI:10.1039/c3gc41403e |
| [31] |
Y. Zhang, K.B. Teuscher, H. Ji, Chem. Sci. 7 (2016) 2111-2118. DOI:10.1039/C5SC03640B |
| [32] |
H.Y. Song, F. Xiao, J. Jiang, et al., Chin. Chem. Lett. 34 (2023) 108509. DOI:10.1016/j.cclet.2023.108509 |
| [33] |
Q. Zhang, K. Liang, C. Guo, Angew. Chem. Int. Ed. 61 (2022) e202210632. DOI:10.1002/anie.202210632 |
| [34] |
K. Yamamoto, M. Kuriyama, O. Onomura, Curr. Opin. Electrochem. 28 (2021) 100714. DOI:10.1016/j.coelec.2021.100714 |
| [35] |
J.C. Siu, N. Fu, S. Lin, Acc. Chem. Res. 53 (2020) 547-560. DOI:10.1021/acs.accounts.9b00529 |
| [36] |
K.J. Jiao, Z.M. Li, X.T. Xu, et al., Org. Chem. Front. 5 (2018) 2244-2248. DOI:10.1039/c8qo00507a |
| [37] |
X. Wang, S. Wu, Y. Zhong, et al., Chin. Chem. Lett. 34 (2023) 107537. DOI:10.1016/j.cclet.2022.05.051 |
| [38] |
T.V. Münchow, S. Dana, Y. Xu, B. Yuan, L. Ackermann, Science 379 (2023) 1036-1042. DOI:10.1126/science.adg2866 |
| [39] |
H.Y. Zhou, H.T. Tang, W.M. He, Chin. J. Catal. 46 (2023) 4-10. DOI:10.1016/S1872-2067(22)64197-4 |
| [40] |
X.L. Lai, M. Chen, Y. Wang, J. Song, H.C. Xu, J. Am. Chem. Soc. 144 (2022) 20201-20206. DOI:10.1021/jacs.2c09050 |
| [41] |
P. Zhou, W. Li, J. Lan, T. Zhu, Nat. Commun. 13 (2022) 3827. DOI:10.1038/s41467-022-31453-7 |
| [42] |
C.H. Ou, Y.M. Pan, H.T. Tang, Sci. China Chem. 65 (2022) 1873-1878. DOI:10.1007/s11426-022-1360-3 |
| [43] |
A. Vega-Peñaloza, S. Paria, M. Bonchio, L. Dell' Amico, X. Companyó, ACS Catal. 9 (2019) 6058-6072. DOI:10.1021/acscatal.9b01556 |
| [44] |
L. Zhu, D. Wang, Z. Jia, et al., ACS Catal. 8 (2018) 5466-5484. DOI:10.1021/acscatal.8b01263 |
| [45] |
F. An, B. Maji, E. Min, A.R. Ofial, H. Mayr, J. Am. Chem. Soc. 142 (2020) 1526-1547. DOI:10.1021/jacs.9b11877 |
| [46] |
A.E. Allen, D.W.C. Macmillan, J. Am. Chem. Soc. 132 (2010) 4986-4987. DOI:10.1021/ja100748y |
| [47] |
B. List, R.A. Lerner, C.F. Barbas Ⅲ, J. Am. Chem. Soc. 122 (2000) 2395-2396. DOI:10.1021/ja994280y |
| [48] |
E. Arceo, I.D. Jurberg, A. Álvarez-Fernández, P. Melchiorre, Nat. Chem. 5 (2013) 750-756. DOI:10.1038/nchem.1727 |
| [49] |
S. Mondal, F. Dumur, D. Gigmes, et al., Chem. Rev. 122 (2022) 5842-5976. DOI:10.1021/acs.chemrev.1c00582 |
| [50] |
S. Mukherjee, J.W. Yang, S. Hoffmann, B. List, Chem. Rev. 107 (2007) 5471-5569. DOI:10.1021/cr0684016 |
| [51] |
D.W.C. Macmillan, Nature 455 (2008) 304-308. DOI:10.1038/nature07367 |
| [52] |
Z.H. Wang, P.S. Gao, X. Wang, et al., J. Am. Chem. Soc. 143 (2021) 15599-15605. DOI:10.1021/jacs.1c08671 |
| [53] |
N. Fu, L. Li, Q. Yang, S. Luo, Org. Lett. 19 (2017) 2122-2125. DOI:10.1021/acs.orglett.7b00746 |
| [54] |
F.Y. Lu, Y.J. Chen, Y. Chen, et al., Chem. Commun. 56 (2020) 623-626. DOI:10.1039/c9cc09178e |
| [55] |
J. Jiang, K.L. Wang, X. Li, et al., Chin. Chem. Lett. 34 (2023) 108699. DOI:10.1016/j.cclet.2023.108699 |
| [56] |
X.H. Ho, S. Mho, H. Kang, H.Y. Jang, Eur, J. Org. Chem. 2010 (2010) 4436-4441. DOI:10.1002/ejoc.201000453 |
| [57] |
K.L. Jensen, P.T. Franke, L.T. Nielsen, K. Daasbjerg, K.A. Jørgensen, Angew. Chem. Int. Ed. 49 (2010) 129-133. DOI:10.1002/anie.200904754 |
| [58] |
M.D. Palkowitz, G. Laudadio, S. Kolb, et al., J. Am. Chem. Soc. 144 (2022) 17709-17720. DOI:10.1021/jacs.2c08006 |
| [59] |
K. Zhou, Y. Yu, Y.M. Lin, Y. Li, L. Gong, Green Chem. 22 (2020) 4597-4603. DOI:10.1039/d0gc00262c |
| [60] |
J.F. Van Humbeck, S.P. Simonovich, R.R. Knowles, D.W.C. Macmillan, J. Am. Chem. Soc. 132 (2010) 10012-10014. DOI:10.1021/ja1043006 |