 2024, Vol. 35
2024, Vol. 35 


b School of Chemistry and Chemical Engineering, Shandong University, Ji'nan 250100, China;
c Department of Chemistry, Faculty of Science, King Khalid University, Abha 61413, Saudi Arabia
 Shehla Khalid received her Bachelor's degree from Government College University, Faisalabad. She completed her master's degree under the supervision of Prof. Nasir Rasool from the same institute. Her research interest focuses on transition metal catalyzed reactions, chemistry of heterocycles, photo/electrochemical reactions, bio-active molecules synthesis, coupling reactions, inert bonds activation and functionalization;
Shehla Khalid received her Bachelor's degree from Government College University, Faisalabad. She completed her master's degree under the supervision of Prof. Nasir Rasool from the same institute. Her research interest focuses on transition metal catalyzed reactions, chemistry of heterocycles, photo/electrochemical reactions, bio-active molecules synthesis, coupling reactions, inert bonds activation and functionalization; |
 Muhammad Bilal received his Bachelor's and Master's degrees from Government College University, Faisalabad, under the supervision of Prof. Nasir Rasool. Then, he joined Higher Education Department Pakistan as a Lecturer of Chemistry. Now, he is doing Ph.D. as a CSC fellow under the supervision of Prof. Yu-Feng Liang at Shandong University, China. Currently, his doctoral research focuses on cross coupling reactions using electrochemistry or earth-abundant transition metals and ring-opening reactions;
Muhammad Bilal received his Bachelor's and Master's degrees from Government College University, Faisalabad, under the supervision of Prof. Nasir Rasool. Then, he joined Higher Education Department Pakistan as a Lecturer of Chemistry. Now, he is doing Ph.D. as a CSC fellow under the supervision of Prof. Yu-Feng Liang at Shandong University, China. Currently, his doctoral research focuses on cross coupling reactions using electrochemistry or earth-abundant transition metals and ring-opening reactions; |
 Nasir Rasool was born in Chichawatni, Sahiwal, Pakistan. He received his MSc. degree from Bahauddin Zakariya University Multan. Later he joined Prof Dr Peter Langer at the institute of Chemistry, University of Rostock, Germany as PhD research fellow. Then he joined Government College University Faisalabad as an Assistant Professor. He has published more than 200 research articles and supervised number of graduate, post graduate and PhD students. He received research productivity awards from Pakistan council of science and technology several times. His-current research focuses on synthesis of small drug molecules, cross-coupling reactions and synthesis of organic solar cells and theoretical studies. Now, he is working as a Professor of Chemistry at Department of Chemistry GC University, Faisalabad, Pakistan;
Nasir Rasool was born in Chichawatni, Sahiwal, Pakistan. He received his MSc. degree from Bahauddin Zakariya University Multan. Later he joined Prof Dr Peter Langer at the institute of Chemistry, University of Rostock, Germany as PhD research fellow. Then he joined Government College University Faisalabad as an Assistant Professor. He has published more than 200 research articles and supervised number of graduate, post graduate and PhD students. He received research productivity awards from Pakistan council of science and technology several times. His-current research focuses on synthesis of small drug molecules, cross-coupling reactions and synthesis of organic solar cells and theoretical studies. Now, he is working as a Professor of Chemistry at Department of Chemistry GC University, Faisalabad, Pakistan; |
 Muhammad Imran completed his graduation in Chemistry from the University of Agriculture, Faisalabad, Pakistan and obtained his PhD degree from International Center for Chemical and Biological Science, University of Karachi, Pakistan in 2009. Then he joined Higher Education Department Lahore, Pakistan as Assistant Professor in 2009. He worked as a post-doc researcher at University of Sao Paulo Brazil in 2011 and at University of Bristol UK in 2017. His-research interest focused on biologically active natural, synthetic products, functional nanomaterials, and first principle investigations. At present, he is working as Associate professor of organic chemistry at King Khalid University, Abha, Saudi Arabia
Muhammad Imran completed his graduation in Chemistry from the University of Agriculture, Faisalabad, Pakistan and obtained his PhD degree from International Center for Chemical and Biological Science, University of Karachi, Pakistan in 2009. Then he joined Higher Education Department Lahore, Pakistan as Assistant Professor in 2009. He worked as a post-doc researcher at University of Sao Paulo Brazil in 2011 and at University of Bristol UK in 2017. His-research interest focused on biologically active natural, synthetic products, functional nanomaterials, and first principle investigations. At present, he is working as Associate professor of organic chemistry at King Khalid University, Abha, Saudi Arabia |
Chemists have been fascinated by the potential of visible light in organic synthesis since its early days [1-4]. It offers a renewable energy source, making photochemistry inherently connected to sustainability. The synthetic potential of photochemistry is equally alluring, unlocking unique reaction pathways not accessible through conventional ground-state reactivity [5-7]. However, until recently, photochemistry faced limitations due to specialized light apparatus and scattered absorption properties of organic molecules. The advent of photoredox catalysis in the past decade has revolutionized photochemical reactions, making them more widely applicable and efficient. These advancements in synthetic chemistry hold promise for industries like pharmaceuticals and agrochemicals, enabling expanded chemical space, improved route efficiency, and safer processes. Embracing novel technologies can simplify synthesis, unlock diverse chemical possibilities, and create novel biologically active compounds [8-14].
The demand for environmental protection and sustainable practices drives the pursuit of green and eco-friendly synthetic strategies for organic transformations. Visible light-promoted photoredox catalysis has emerged as an attractive approach, leveraging visible light as a mild and powerful tool for activating organic molecules through single-electron transfer (SET) processes [1,15-24]. With renowned researchers contributing seminal studies, visible-light photoredox catalysis continues attracting significant attention and driving innovations in organic chemistry [25-36].
In their excited states, molecular chromophores frequently display advantageous redox characteristics. A charge-separated electron–hole pair, known as an electrically excited state, is created when a photon promotes an electron from the HOMO to LUMO. This excited state can either donate its high-energy electron to a suitable acceptor or accept an electron from a suitable electron donor if it lasts long enough for future intermolecular reactions. As a result of possessing an electron−hole pair, an excited photocatalyst can participate in both oxidative and reductive chemistry, making it a more potent reductant and oxidant than its ground-state counterpart.
Developments in synthetic chemistry have profound implications for both the agrochemical and pharmaceutical industries. They offer the potential to expand the accessible chemical space, improvement in route effectiveness, and facilitate the adoption of cost-effective and safer methodologies [37,38]. While these industries tend to favor classical and powerful transformations, adopting new synthetic methodologies can be slow. Nonetheless, embracing new technologies can lead to more straightforward synthesis, broadening chemical space and enabling the synthesis of pharmaceuticals. The acceptance of such methods is generally guided by various criteria, which include the availability of starting materials, catalysts, reagents, specialized equipment, and the overall practical feasibility of the approach [39].
At the heart of this progress is the utilization of light to access reactive intermediates, enabling the development of novel bond-forming protocols not easily achievable under thermal conditions. Photoredox catalysis stands out for its ability to operate under very mild reaction conditions, which making it suitable for late-stage functionalization and more functionalized compounds [40-45]. Moreover, photoredox catalysis grants chemists the opportunity to utilize safer, cost-effective, and environmentally friendly reagents while enabling novel disconnections that can lead to enhanced synthetic routes and the development of valuable intellectual property [46]. The categorization of photochemical reactions into allylic additions, cyclization, reductive cross-coupling, C—H activation, ring opening, oxidative cross-coupling, dehydrogenation, desulphonation, and decarboxylation represents a systematic and comprehensive approach to harness the energy of light-induced transformations for the synthesis of pharmaceutically relevant scaffolds and compounds. This categorization not only reflects the richness and diversity of photochemical methods but also underscores their utility in addressing the intricate challenges of synthesizing biologically active molecules. As a result, this systematic classification provides researchers with a broad and nuanced framework to navigate the complex landscape of photochemical synthesis for pharmaceutical applications. This review covers the five-year overview of the potential photoredox methodologies which have the potential for drug synthesis or late-stage functionalization.
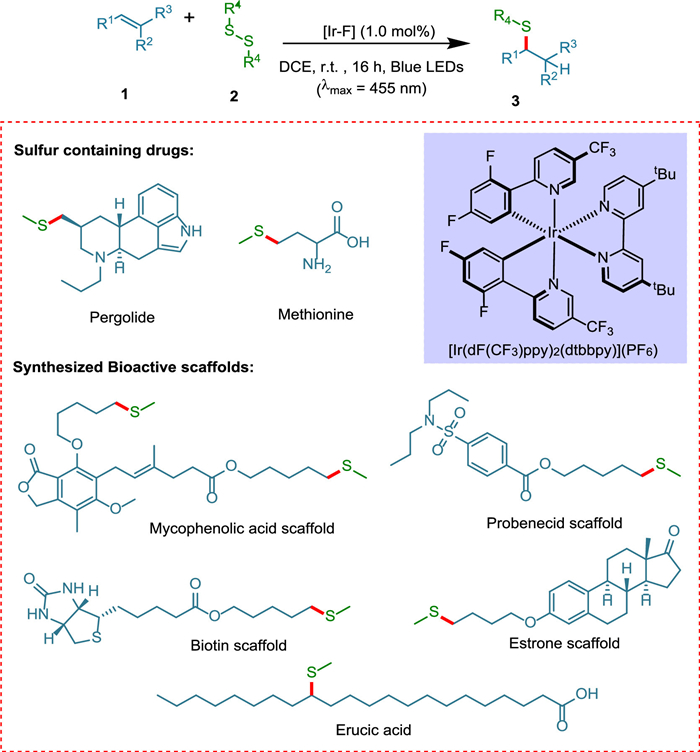
2. Allylic additions reactionsAddition reaction finds its worth in its variety of approaches, e.g., Markovnikov-type radical addition using radical hydrogen [47], conjugate addition (1,2-, 1,4-, and 1,6-) controls stereo and regiochemistry of the product by introducing chiral center [48], cycloaddition (inter-, intra-) [49,50], allylic addition [51] and aldol addition reaction [52]. The addition reaction is one of the most important steps in polymerization, and recently it has been performed in the presence of light and photocatalyst Ru(bpy)3Cl2 [53]. In 2018, Rossolini et al. synthesized α-amino-γ-lactam from a photocatalyzed addition reaction between imine and electrophilic olefin moiety [54]. In 2020, Wang et al. opted for a nanocage-confined approach to photocatalyzed synthesis of cyclobutane derivatives via cycloaddition reaction [55]. In 2022, Vijayakrishnan et al. synthesized (±)-rolipram drug in the presence of CTF-2 as a photocatalyst for decarboxylative conjugate addition [56]. The methylthioether scaffolds are involved in various metabolic processes and may be found in key pharmaceuticals such as methionine and pergolide because thiol-ene click reaction in the presence of a photocatalyst occurs under mild reaction conditions and is frequently employed in bioconjugate molecules, polymer chemistry, and drug manufacturing [51]. Lately, UV rays have been commonly utilized to generate thiyl radicals [57]. Though, the limited substrate scope, expensive UV irradiation setups, and the creation of massive waste have been limiting considerations [58]. Teders et al. proposed a chemoselective anti-Markovnikov hydroalkyl/aryl thiolation of alkenes and alkynes, opening the door for the hydromethylthiolation of disulfides via triplet–triplet EnT (Scheme 1) [51]. These are generally partitioned into three classes: alkenes that have been activated undergo E/Z isomerization [59], [2 + 2] photocycloadditions [60,61], and transition metal complex sensitization [62]. Alkenes can be hydromethylated with many functional groups, such as ketones, aldehydes, epoxides, alcohols, nitriles, sulfonamides, amides, heterocycles, and esters. This methodology yields products of 42% to 85%.

|
Download:
|
| Scheme 1. The hydromethylthiolation of disulfides. | |
{kind=link}
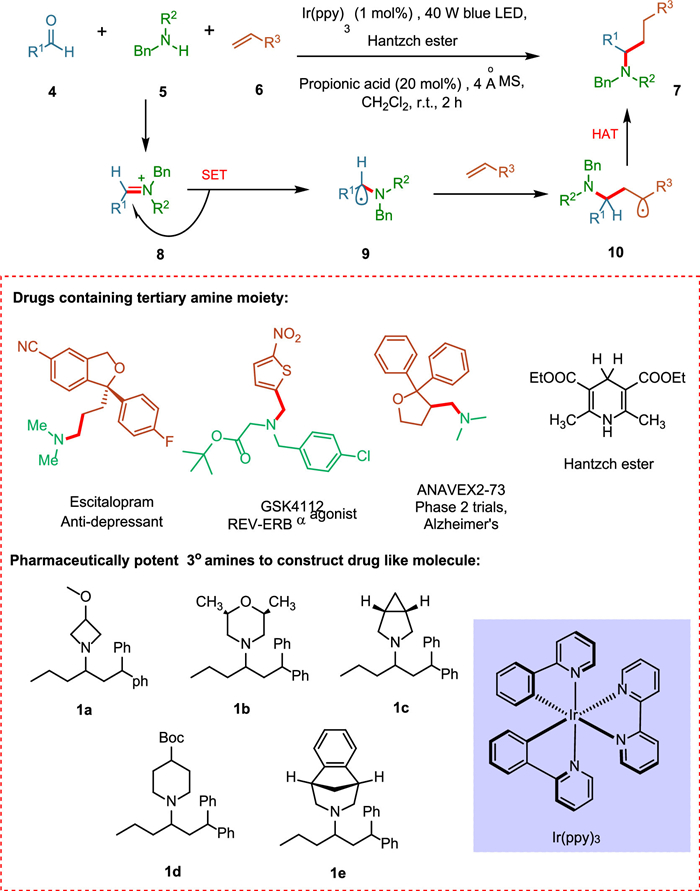
Tertiary aliphatic amines 7 are highly effective pharmaceutical agents due to their physiological properties and ability to disrupt natural neurotransmission pathways [63]. These applications cover various biological treatments, from neuroimpaired diseases [64] to metabolic issues [65]. The Buchwald–Hartwig amination [66] and olefin-hydroamination [67,68] methods have limitations in forming complex alkylamines in the novel transition-metal-catalyzed amine synthesis strategies [69,70]. Trowbridge et al. recommended that a photoexcited photocatalyst could simultaneously intervene in SET to synthesize alkylated iminium ion, giving rise to the required α-amino radical under mild conditions. The core of this approach lies in the regiospecificity of the newly synthesized α-amino radical. The iminium ion 8, is formed by the combination of alkyl-substituted carbonyls 4 and secondary alkylamines 5, undergoes SET resulting α-amino radical 9, which reacts with an alkene 6, forming sp3 hybridized carbon-carbon bond 10 (Scheme 2) [45].

|
Download:
|
| Scheme 2. Synthesis of tertiary aliphatic amines. | |
{kind=link}
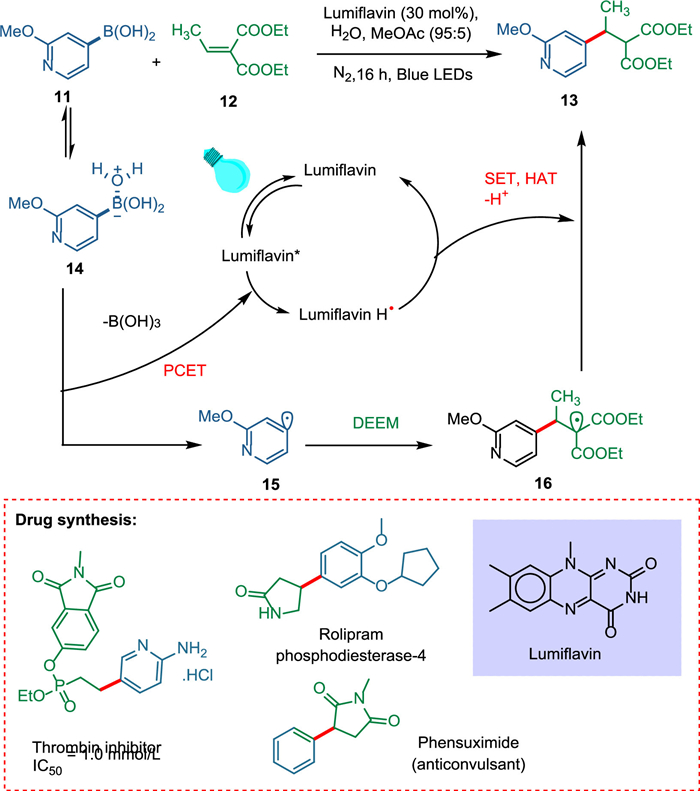
Chilamari et al. proposed an aqueous technique that uses a bioinspired flavin photocatalyst to convert a broad range of boronic acids to carbon radicals which are employed to deliver different kinds of alkylated pharmaceutical compounds. Water coordinates with the boronic acid 11, which results in the aqua-boryl complex 14. The excited PC lumiflavin removes a proton and an electron from 14. The fragmentation of the intermediate 14 forms the heteroaryl radical 15. The former is quickly captured by adding open-shell conjugates to diethyl ethylidene malonate 12 to produce the α-malonyl radical 16, which is then reduced to produce the required alkylated product 13, and the photocatalyst is regenerated (Scheme 3) [71].

|
Download:
|
| Scheme 3. A bioinspired flavin photocatalyst employed in boonic acid conversion to carbon radicals. | |
{kind=link}
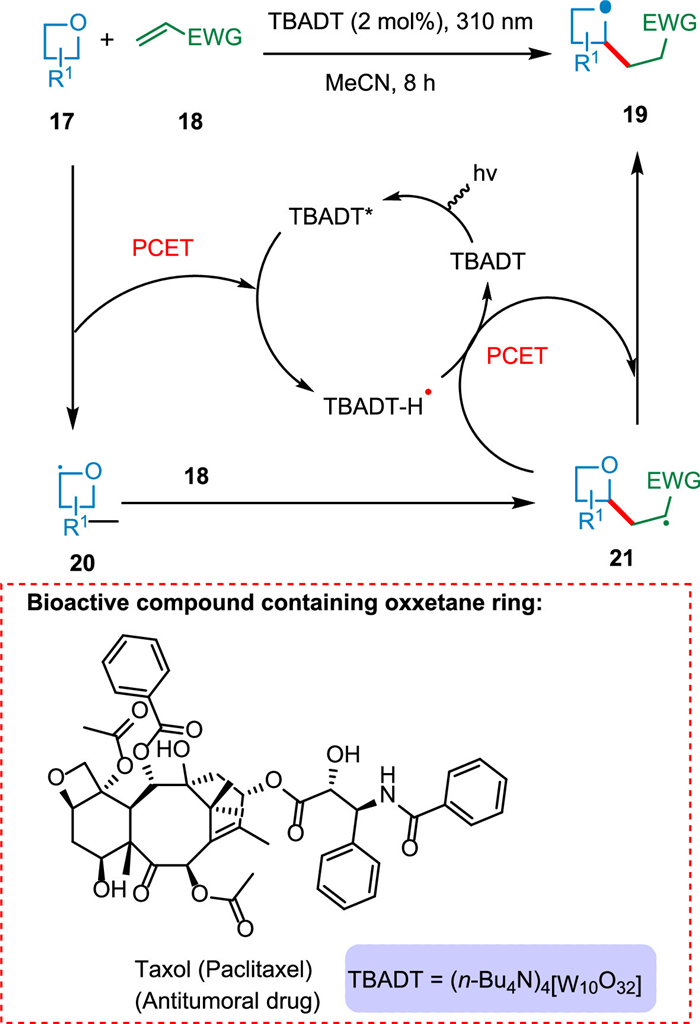
The oxetane ring is found in several significant natural products and pharmaceuticals, such as taxol (paclitaxel), an antitumoral agent [72]. Ravelli et al. used decatungstate photocatalysis to achieve C(sp3)-H activation in oxetanes 17 selectively at a second position under mild conditions. The resultant α-oxy radical 20 is attacked by electrophilic alkene 18 and produces 2-substituted oxetanes 19 (Scheme 4) [73].

|
Download:
|
| Scheme 4. Oxetane ring activation. | |
{kind=link}
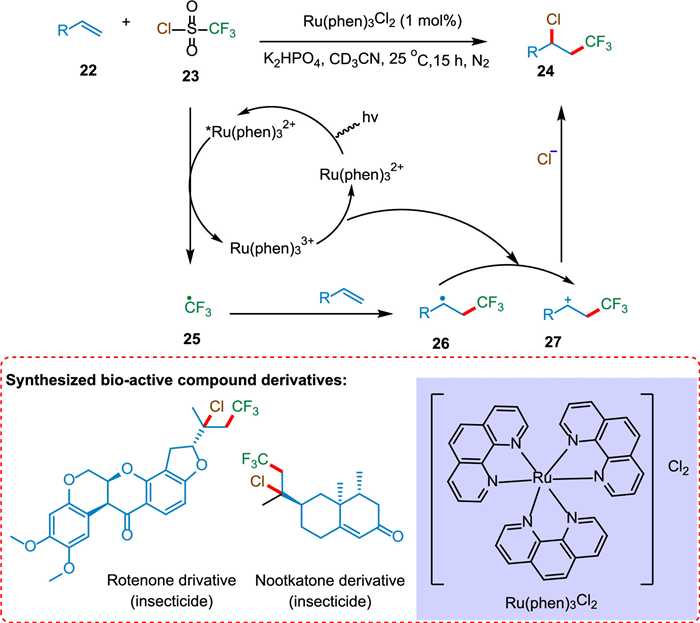
Oh et al. reported the photoredox catalysis of vicinal chlorotrifluoromethylation of alkene. The SET from photoexcited PC *Ru(Phen)3Cl2 to CF3SO2Cl (23) forms the CF3 radical 25, which reacts with an alkene to form the radical intermediate 26. The SET from 26 to oxidized PC Ru(Phen)33+ regenerates ground state PC and creates cationic intermediate 27, which was captured by Cl− to form the product 24 (Scheme 5). Numerous terminal and internal alkenes underwent vicinal chlorotrifluoromethylation to produce their derivatives. In order to produce the desired results, biologically active chemicals were used under the conditions [74].

|
Download:
|
| Scheme 5. The photoredox catalysis of vicinal chlorotrifluoromethylation of alkene. | |
{kind=link}
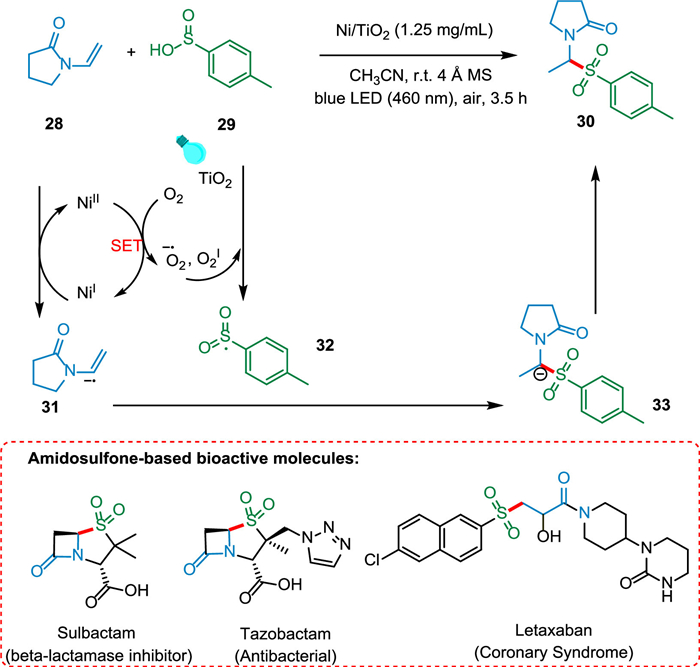
The increased interest in single-atom photocatalysis is evident as it is a significant method of organic transformation, with efficacy influenced by catalyst design. Yang et al. reported a visible light mediated approach that uses a single-atom photocatalyst (Ni/TiO2) to sulfonate enamides producing amidosulfones and β-propionamidosulfones. The singlet oxygen IO2 oxidizes p-methylbenzenesulfonic acid 29, generating the sulfone radical and molecular oxygen is reduced by remaining photo-generated electrons forming O2 radical and 1O2. The remaining electrons will be transferred to NiII to produce NiI, which can then be further oxidized by enamide 28 to produce NiII and radical anion intermediate 31, which is attacked by a sulfone radical 32 to produce carbanion intermediate 33. The desired product, α-amidosulfones 30, can be produced by protonating carbanion intermediate 33 (Scheme 6). This single-atom photocatalysis-based synthesis approach has excellent functional group tolerance, is easily scaled up, and is highly recyclable [75].

|
Download:
|
| Scheme 6. The synthesis of amidosulfones and β-propionamidosulfones. | |
{kind=link}
3. Cyclization reactions
In 2023, Lu et al. synthesized benzo[e]imidazo[1,5-c][1,2,3]oxathiazine 5,5-dioxide derivatives by cyclization reaction using the combination of heterogeneous semiconductor photocatalyst (WSe2) and homogeneous redox catalyst (ferrocene) [76]. Ji et al. proposed a mild photo-induced approach to synthesize naphthoselenazol-2-amines at ambient temperature in the presence of air as an oxidant and water as solvent without employing an exogenous photosensitizer or additive [77].
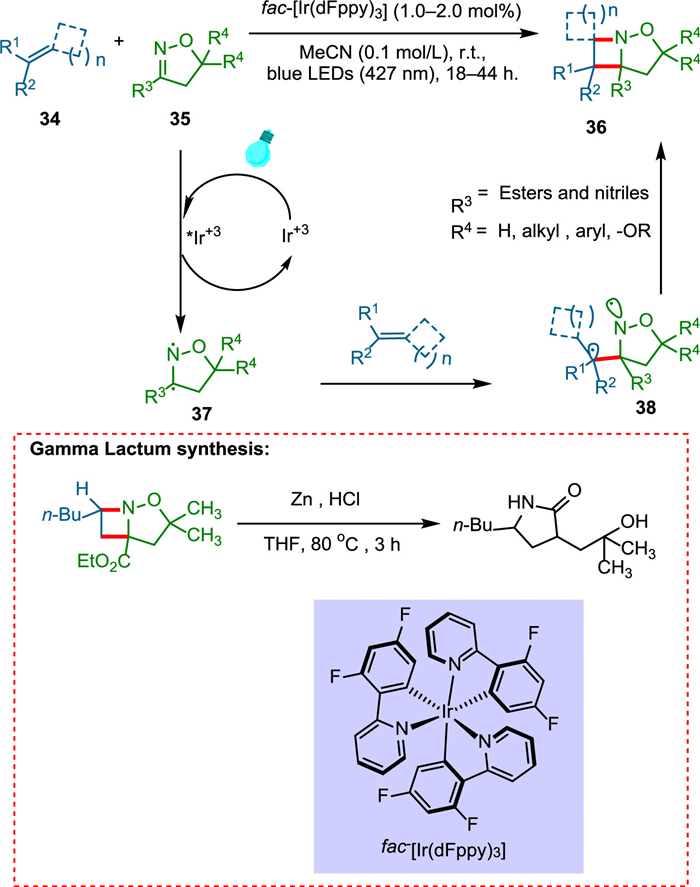
Four-membered azetidine heterocycles are important scaffolds in drug development [78,79]. Maruoka and coworkers revealed that N-aryl sulfonyl imines successfully participate in the [2 + 2] photocycloaddition reaction, yielding protected azetidines [80]. Unfortunately, this technique is confined to aromatic imines and styrenes because the singlet-state between the reactants is required. Becker et al. discovered an intermolecular aza Patern-Büchi reaction initiated by visible light that makes the use of unusually high triplet state energy of oximes, which is easily accessible via photocatalyst of iridium complex, allowing cycloaddition with a wide variety of olefins. Visible light excites the photocatalyst, which transfers energy effectively to 35 to form triplet-state isoxazoline 37, which undergoes stepwise cycloaddition with alkene 34 to give rise to major diaseteromer product biradical intermediate 38, in which there is a minimal steric hindrance between the alkene moiety and the oxazole moiety 35 backbone (Scheme 7). This method enables the formation of azetidines derivatives, which are highly functionalized, from easily accessible starting materials. It is distinguished by its operational simplicity, mild conditions, and broad substrate scope of olefins which could be formerly transformed via UV-light only. Additionally, the utilization of isoxazoline carboxylate is advantageous owing to the formation of unprotected azetidines, which used to be challenging to obtain [81].

|
Download:
|
| Scheme 7. The intermolecular aza Patern-Büchi reaction of oxime with olefins. | |
{kind=link}
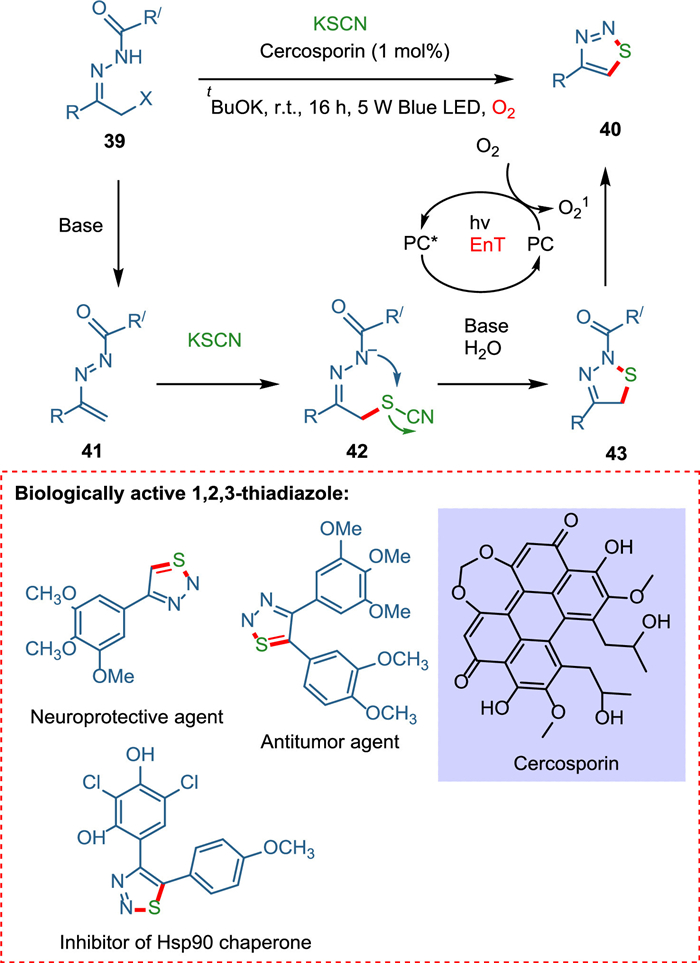
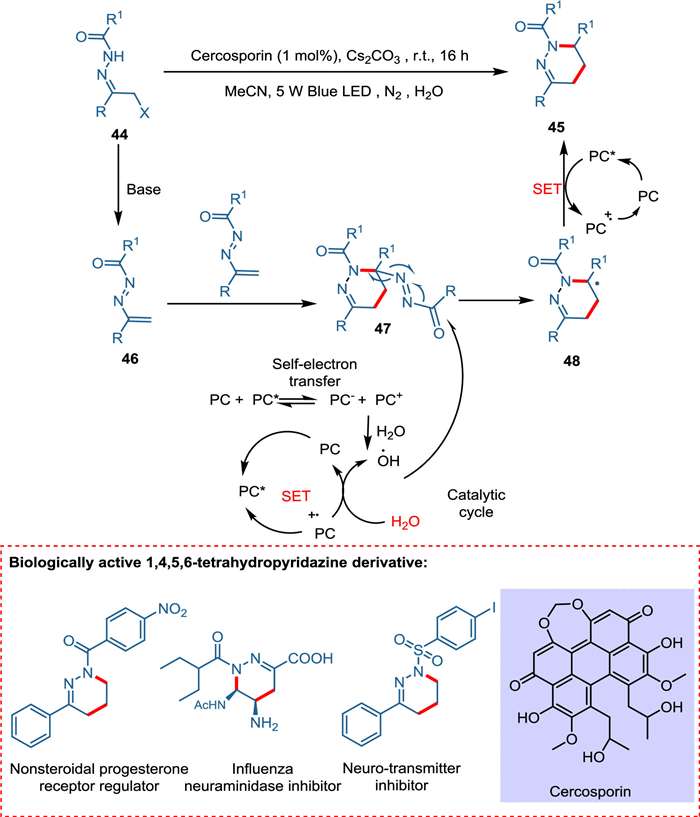
Traditional organic synthesis methods mainly rely on thermal cycloaddition reactions; hence, novel methodologies for forming N-heterocycles under moderate conditions are needed. Zhang et al. devised a novel approach for constructing nitrogen-containing heterocycles under mild conditions by employing cercosporin, a perylenequinonoid derivative, as a metal-free, cost-effective, and green photocatalyst. The [4 + 1] annulation of azoalkenes in the presence of photocatalyst yields 1,2,3-thiadiazoles using thiocyanate salt while photocatalyzed [4 + 2] cyclodimerization of azoalkenes yields 1,4,5,6-tetrahydropyridazines. Base extracts hydrogen from α-halo hydrazone 39, releasing the halo group and forming azoalkene 41. The synthesis of 1,2,3-thiadiazoles starts when azoalkene 41 reacts with thiocyanate forming intermediate 42, which transforms into N-acylated thiadiazole 43 and cyano moiety being hydrolyzed [82,83]. The former is aromatized [84] via IO2 [85], yielding 1,2,3-thiadiazole 40 (Scheme 8). While the formation of 1,4,5,6-tetrahydropyridazines is initiated when azoalkene 47 gives pyridazine 48 via a radical pathway. The OH radical, which is formed by PC self-electron transfer attacks at carbonyl carbon of 48, yielding radical intermediate 49, which via SET followed by protonation, forms the product (1,4,5,6-tetrahydropyridazine) (Scheme 9) [86].

|
Download:
|
| Scheme 8. The cercosporin catalyzed [4 + 1] annulation of azoalkenes yielding 1,2,3-thiadiazoles. | |
{kind=link}

|
Download:
|
| Scheme 9. The cercosporin catalyzed [4 + 2] cyclodimerization of azoalkenes yields 1,4,5,6-tetrahydropyridazines. | |
{kind=link}
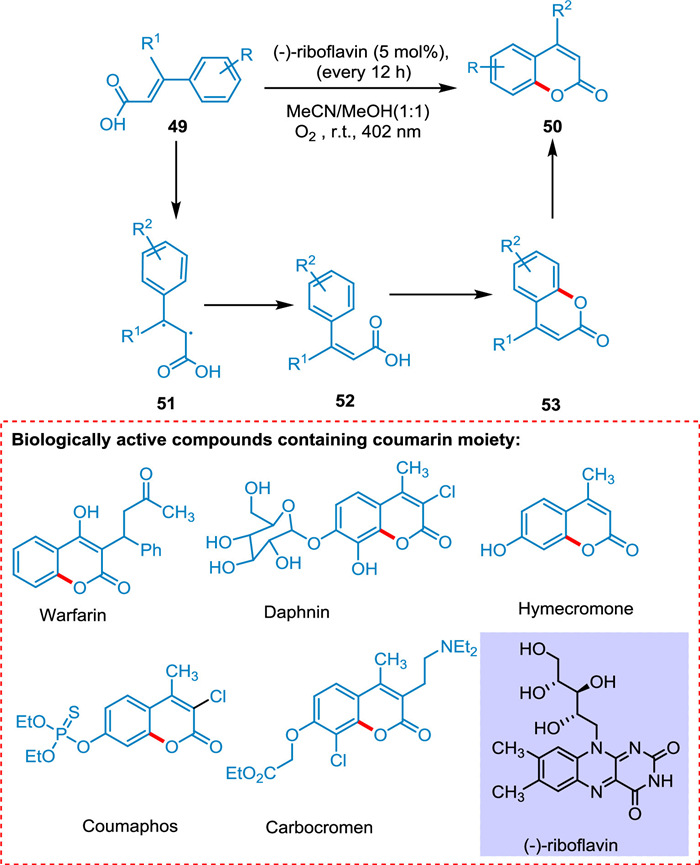
Metternich and Gilmour reported a direct coumarin synthesis from E-cinnamic acids. This biomimetic method does not need the aryl ring to be prefunctionalized before cyclization. It is possible to enable isomerization and cyclization consecutively via energy transfer (ET) and SET activation routes of (-)-riboflavin by leveraging the two separate photochemical activation mechanisms. The basic mechanistic idea, that blends ET followed by SET, integrates proven "one catalyst, two reactions" methods. A biradical intermediate 51 converts (E)-cinnamic acid 49 to (Z)-cinnamic acid 52. This isomerization arises through photocatalysis. The SET process converts (Z)-cinnamic acid 52 to coumarin 50, and reduced RFH2 is released, which is oxidized to (-)-riboflavin, completing the catalytic cycle (Scheme 10) [87].

|
Download:
|
| Scheme 10. A direct coumarin synthesis from E-cinnamic acids. | |
{kind=link}
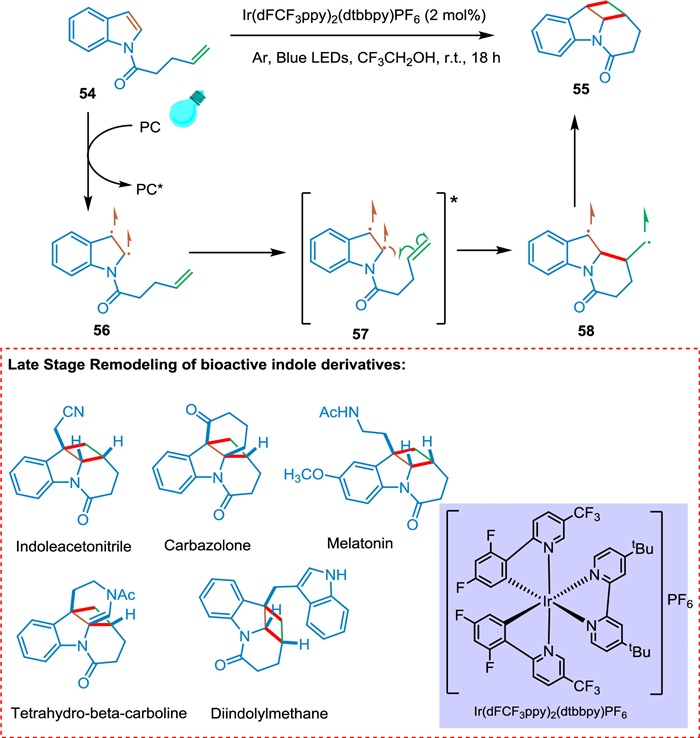
Numerous alkaloid-based natural products and pharmaceuticals contain polycyclic indoline scaffolds, and significant effort has been made in recent years to synthesize them [88,89]. High energies of the excited state favor photoinduced dearomative [2 + 2] cycloaddition of indole derivatives linked to alkene at the N1 position has been thought to be energetically impractical. Zhang et al. explained visible-light favored [2 + 2] cycloaddition with accompanied dearomatization of indole derivatives linked to olefins at N1 position yielding cyclobutane-fused indoline-based polycycles which have remarkable yields and diastereoselectivities (> 99:1). The indole derivative 54 in the presence of photocatalyst goes converted into biradical 56 via EnT followed by excited triplet state 57 which undergoes intramolecular cyclization between olefin moiety and C2, C3 positions of indole ring forming tricyclic biradical indole derivative 58 with new bridged bond at C2. Radicals combine to form a new bond, and the targeted product 55 is achieved (Scheme 11). The creation of hydrogen bonding between product and solvent decreases the reactant's triplet state energy and significantly increases productivity. Late-stage modification of bioactive indole derivatives serves as an example of the method's application [90].

|
Download:
|
| Scheme 11. Synthesis of cyclobutane-fused indoline-based polycycles. | |
{kind=link}
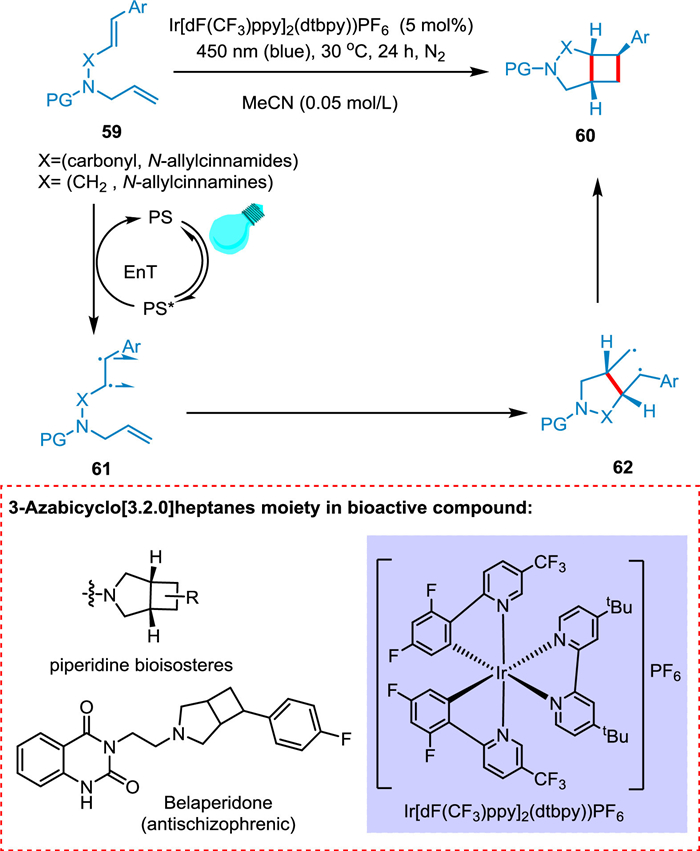
Oderinde et al. put forward that by using photocatalyst (Ir[dF(CF3)ppy]2(dtbpy))PF6, the intramolecular [2 + 2] cycloaddition of N-allylcinnamamines and N-allylcinnamamides is possible. The former is converted into aryl-3-azabicyclo[3.2.0]heptanones while the later into aryl-3-azabicyclo[3.2.0]heptanes. The iridium photosensitizer under milder conditions is effective in stimulating this type of reaction owing to its high triplet EnT and diastereoselectivity. This reaction has broad substrate scope, proven to be tolerated in drug development. Energy transfer from PC to N-allylcinnamamides and N-allylcinnamamines 59 results in a biradical triplet state 61, which undergoes triplet to singlet inversion to form 62 followed by ring-closing to yield corresponding products 60 (Scheme 12) [91].

|
Download:
|
| Scheme 12. The intramolecular [2 + 2] cycloaddition of N-allylcinnamamines and N-allylcinnamamides. | |
{kind=link}
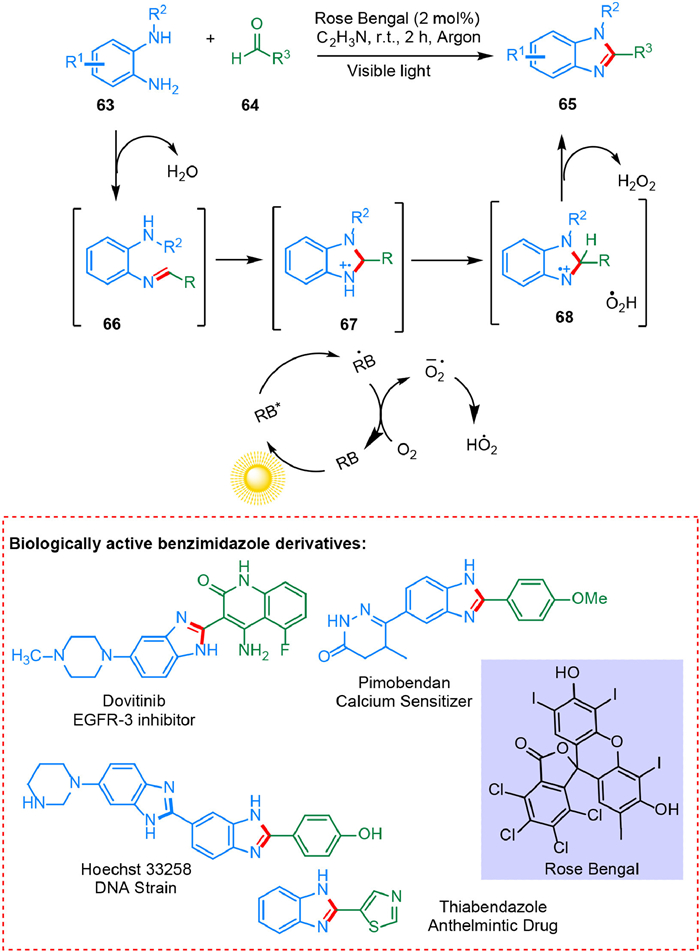
Benzimidazole derivatives exhibit diversified pharmaceutical properties such as antimicrobial [92], anticancer like dovitinib (renal cancer) [93], thiabendazole (anthelmintic activity) [94], pimobendan (vasodilator) [95] and Hoechst stain is used to stain DNA in fluorescence microscopy [96]. Metal-catalyzed condensations need high temperatures, and lingering metal contaminants in the product can result in toxicity in subsequent phases of drug development [97,98]. Kovvuri et al. devised a durable, operationally easy, and highly generic approach for the photocatalytic condensation of o-phenylenediamines and aldehydes in metal-free conditions to produce functionalized benzimidazoles. This method was effective for combining less reactive heterocyclic aldehydes with o-phenylenediamines. Unreactive electron-rich aldehydes are useful in this technique. The rose Bengal can be collected and reused thrice during the catalytic process. The imine 66 is generated by the condensation of 63 and 64. Under visible light irradiation, the excited state of RB can take one electron from imine via a SET. The photo-redox cycle of O2 oxidizes the RB radical anion back to the ground-state RB. The imine radical cation 67 undergoes HAT resulting in 68, and hydrogen abstraction by the hydroperoxyl radical results in the formation of product 65 (Scheme 13) [99].

|
Download:
|
| Scheme 13. Synthesis of functionalized benzimidazoles. | |
{kind=link}
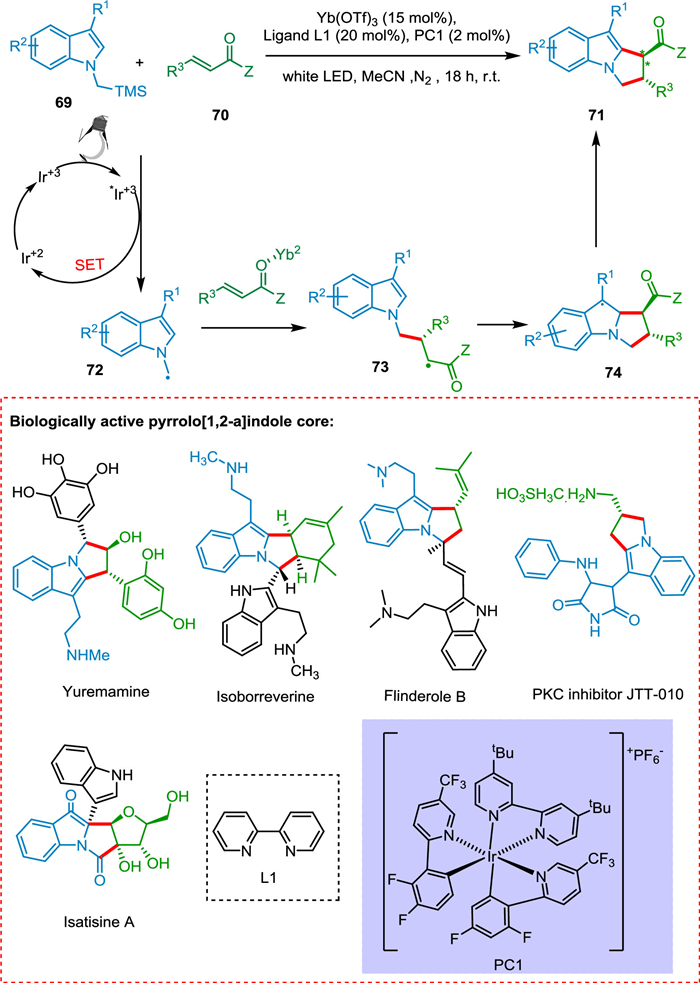
Stephenson's group published the preparation of racemic pyrrolo[1,2-a]indoles using Ru(bpy)32+ as PC [100]. Li described the production of difluoroalkylated pyrrolo[1,2-a]indoles employing the fac-Ir(ppy)3 as a PC [101]. All of these photocatalytic reactions produced a racemic mixture. Sánchez et al. achieved the asymmetric pyrrolo[1,2-a]indoles synthesis in stereoselective fashion through [3 + 2] cycloaddition between indole derivatives 69 and α, β-unsaturated derivatives 70 employing a dual catalytic combination as an IrIII photocatalyst and lewis acid. The former 69 is used to form the α-amino radical 72, which reacts with α, β-unsaturated derivative 70 to form a radical intermediate 73. The homolytic breakage of the carbon-carbon pi bond is followed by a radical combination to form a tricyclic radical intermediate 74, which undergoes HAT to form the targeted product 71 (Scheme 14). Pyrrolo[1,2-a]indole is an important scaffold in drug synthesis [102]. Among them, JTT-010 is an important protein kinase C (PKC) inhibitor [103].

|
Download:
|
| Scheme 14. The asymmetric pyrrolo[1,2-a]indoles synthesis. | |
{kind=link}
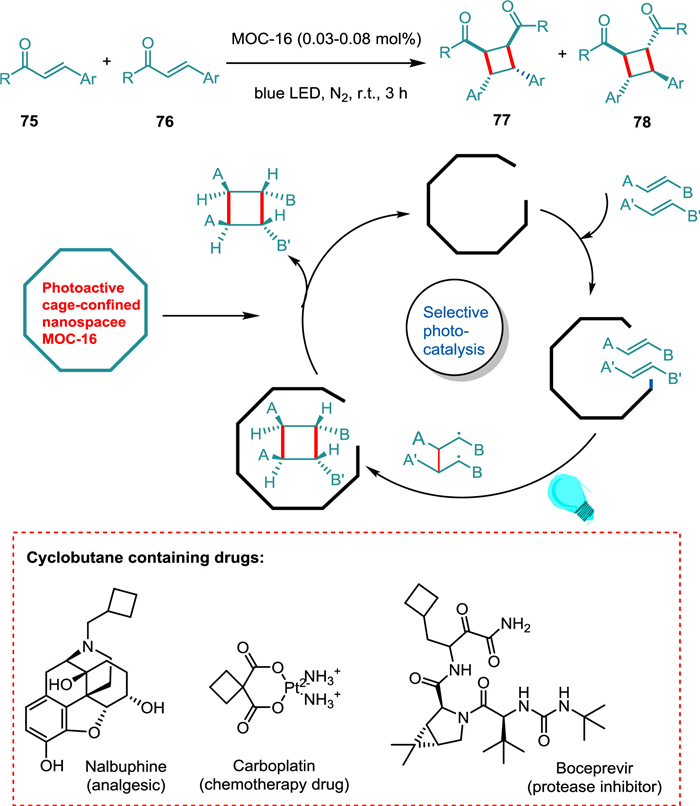
Natural products and bioactive molecules frequently contain cyclobutane scaffolds [104,105], dramatically sparking synthetic interest in building four-membered carbocycles [106,107]. Considering photoinduced [2 + 2] cycloaddition of acyclic enones and cinnamates, Wang et al. devised a practical synthesis process with remarkable selectivity based on the correct design and photoactive cage nano space in a single MOC supramolecular reactor. The cage-confined catalysis makes the homo- and hetero-coupling photoreaction possible, yielding syn-HH cyclobutanes 77 (Scheme 15). A relatively low photocatalyst loading makes broad substrate scope possible. The supramolecular cage regulates selectivity, concentrates the reaction mixture, speeds up reaction time, encourages EnT, and recycles catalytic operation [55].

|
Download:
|
| Scheme 15. The synthesis of cyclobutane scaffolds. | |
{kind=link}
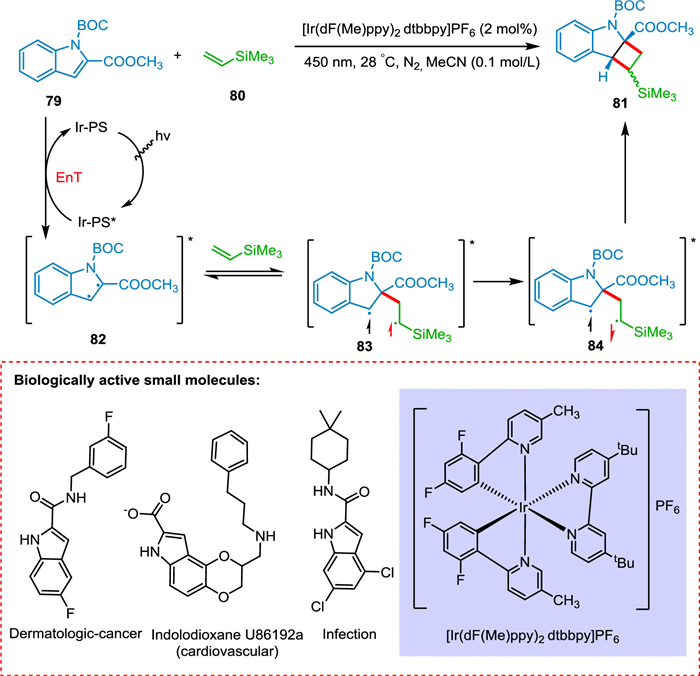
Indole and its bioisosteres are present in drugs approved by the FDA, such as azaindoles, indazole, benzofuran, and benzothiophene [108,109]. Ikeda and colleagues investigated the [2 + 2] cycloaddition between N-benzoylindole and alkenes using UV light [50]. Inevitably, the intermolecular cycloaddition was unsuccessful due to high-intensity UV light, the need for alkene in greater amounts, the limited substrate scope, and undesired homodimerization side-products. Oderinde et al. argued that by studying the relationship between structure and reactivity, N-protected indole-2-carboxyesters and carboxamides could discover those indole derivatives that may undergo [2 + 2] cycloaddition reaction with alkenes derivatives. Oderinde et al. developed a visible-light photocatalytic system for the formation of cyclobutane-fused frameworks through dearomatizing, intermolecular [2 + 2] cycloaddition of indoles and azaindoles with a variety of substituted alkenes, including unactivated alkenes. A triplet-triplet EnT via photocatalyst-photosensitizer (Ir-PS) to N-protected indole-2-carboxyesters 79 initiates the reaction, yielding biradical intermediate (82), which combines with 80 to yield triplet state biradical intermediate 83. The triplet to singlet inversion results in 84 which undergoes ring closure to obtain 81 (Scheme 16). This reaction approach has broad alkene scope giving a high yield. The conversion is highly regio- and diastereoselective [110].

|
Download:
|
| Scheme 16. Visible-light photocatalyzed formation of cyclobutane-fused frameworks. | |
{kind=link}
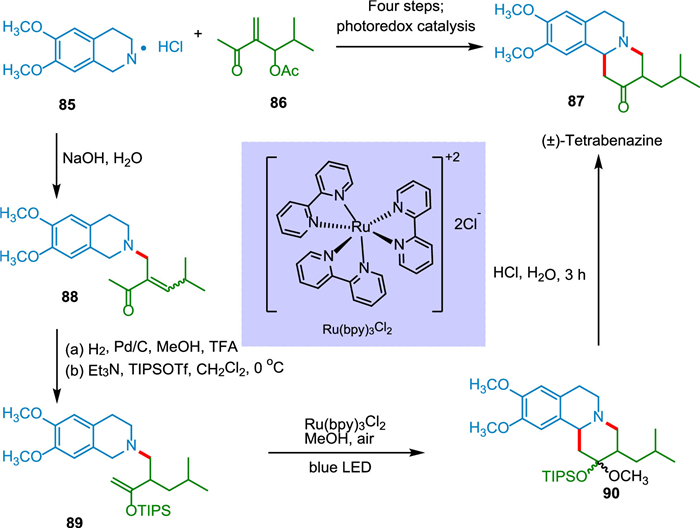
Orgren et al. synthesized (±)-tetrabenazine from readily accessible chemicals in six stages. The cyclization substrate was quickly built using Baylis-Hillman and aza-Michael processes. Photocatalysis results in new ring formation in the presence of visible light, with C—C bond-making via tertiary amine oxidation. The nature of the solvent is crucial in the photoredox cyclization; methanol results in a mixed ketal, while acetonitrile/water results in direct and faster cyclization. When hydrolyzed with NaOH, tetrahydroisoquinoline salt 85 and methyl vinyl ketone 86 facilitate rapid conjugate addition, which is followed by rapid β-elimination of acetate, yielding enone 88. The alkene in enone 88 was selectively reduced to get TIPS enol ether 89 followed by photoredox cyclization with a blue LED and Ru(bpy)3Cl2 catalyst to obtain 90, which was subsequently on acidic hydrolysis to yield (±)-tetrabenazine 87 (Scheme 17) [111]. Tetrabenazine has antipsychotic properties [112].

|
Download:
|
| Scheme 17. Synthesis of (±)-tetrabenazine. | |
{kind=link}
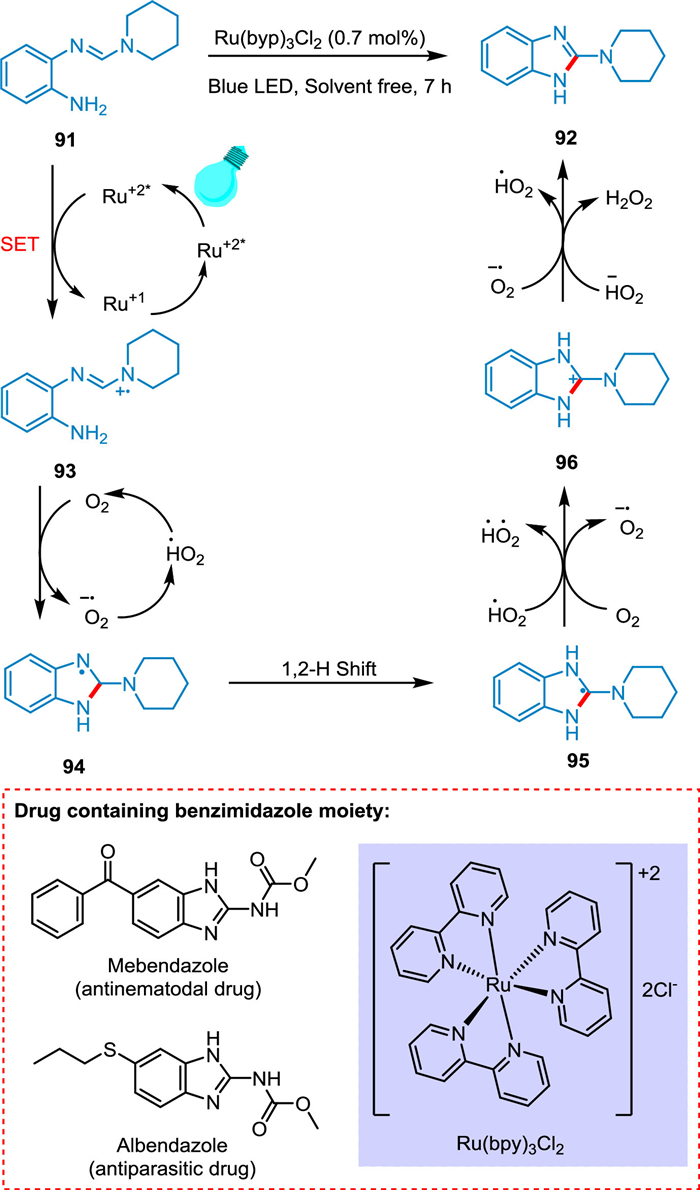
Photocatalytic reactions involve radical chemistry. Radical cyclization is a powerful tool to synthesize products under stereo- and regioselective control. This kind of reaction usually exhibits broad substrate scope and requires milder reaction conditions to build monocyclic and polycyclic complex structures. Cyclization is an intramolecular step involving the addition of radicals to the unsaturated system and trapping the resultant radical via SET [113]. In 2016, Liu et al. synthesized a ring by forming new carbon-carbon bonds in a strained ring system which was earlier difficult to construct. In this strategy, affordable TiO2 was used as a photocatalyst, O2 as an oxidant, and solar light as a light source [114]. In 2019, Li et al. developed a metal and oxidant-free strategy to synthesize cyclization product benzimidazole derivatives using organic dye as a photocatalyst, e.g., fluorescein, and blue LED as a light source [115]. In 2021, Zhuang et al. synthesized γ-lactam derivatives via the HAT/SAT photocatalytic approach through the intramolecular radical cyclization pathway [116]. A key motif in medicinal chemistry, benzimidazole is found in many therapeutic agents [117], including albendazole (antiparasitic), mebendazole (antinematodal) [118] anticonvulsant [119], antimicrobial [120], anti-HIV [121], anti-inflammatory [122], and anticancer [123]. Siddiqui et al. reported a simple and effective strategy for synthesizing the benzimidazole amine hybrid. It involves the interaction of benzimidazole with amine, which can be either cyclic or acyclic, in the presence of photocatalyst Ru(bpy)3Cl2 without using solvent. The oxidation of o-aniline amidines 91 via SET to excited PC (*Ru+2) results in a radical cation species 93. The HAT from 93 to oxygen radical anion followed by intramolecular hetero-cyclization yields aminyl radical 94. The 1,2-H shift of 94 forms radical intermediate 95, which on oxidation produces the corresponding carbocation intermediate 96 and lastly aromatization via deprotonation forming product 92 (Scheme 18). Additionally, the acyclic amines react faster and give the desired product. This approach is an effective replacement for previously described methodologies due to its quick reaction times, outstanding yields, and, most significantly, natural energy source (visible light). This solvent-free approach is aligned with the aim of organic synthesis for generating greener organic reactions due to environmental and energy concerns [124].

|
Download:
|
| Scheme 18. Synthesis of γ-lactam derivatives. | |
{kind=link}
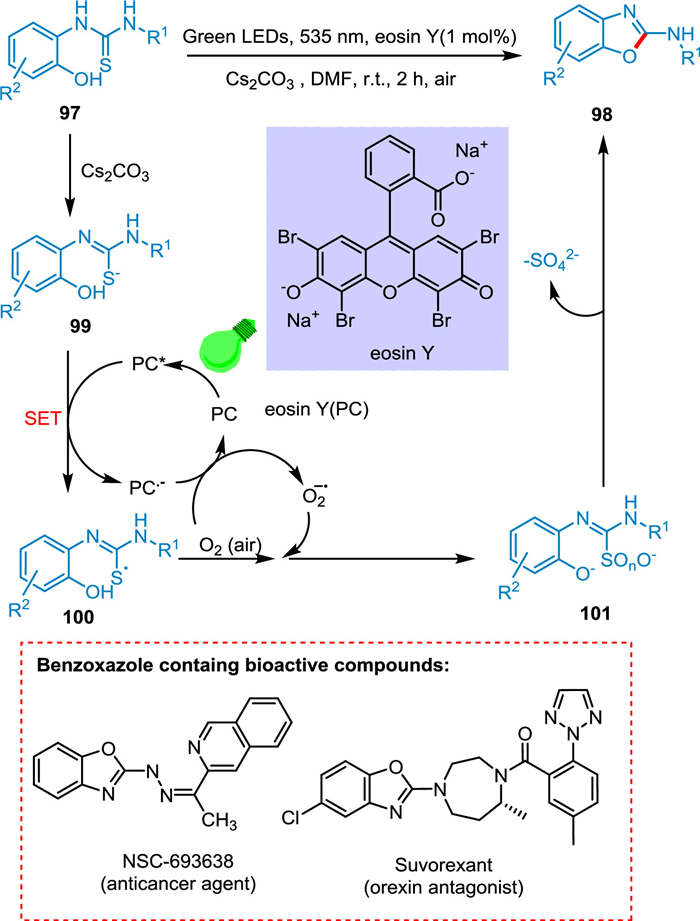
The 2-aminobenzoxazoles are desirable synthetic scaffolds for pharmaceuticals treating inflammatory disorders, HIV, neurodegenerative diseases, insomina, and cancer [125-127]. Yadav et al. reported an effective one-pot procedure for cyclodesulfurization of o-phenolic thioureas to produce 2-aminobenzoxazole derivatives in the presence of light. The base Cs2CO3 abstracts thiolic H from phenolic thioureas 97 to create a more oxidizable sulfur anion 99. Eosin Y (PC) transitions to its excited state (PC*) on absorbing light. The sulfur radical 100 and PC radical anion are produced through a SET between sulfur anion 99 and PC*. The sulfur radical 100 and O2 radical interact to generate an intermediate 101. The targeted products 98 are produced by cyclizing 101 (Scheme 19). The methodology provides a better alternative to the current approaches for the cyclodesulfurization of phenolic thioureas and is carried out as light-mediated process using organo-photocatalyst eosin Y. This strategy makes use of visible light and greener oxidant (air), which is one of the critical characteristics of the current metal-free technique [128].

|
Download:
|
| Scheme 19. Cyclodesulfurization of o-phenolic thioureas. | |
{kind=link}
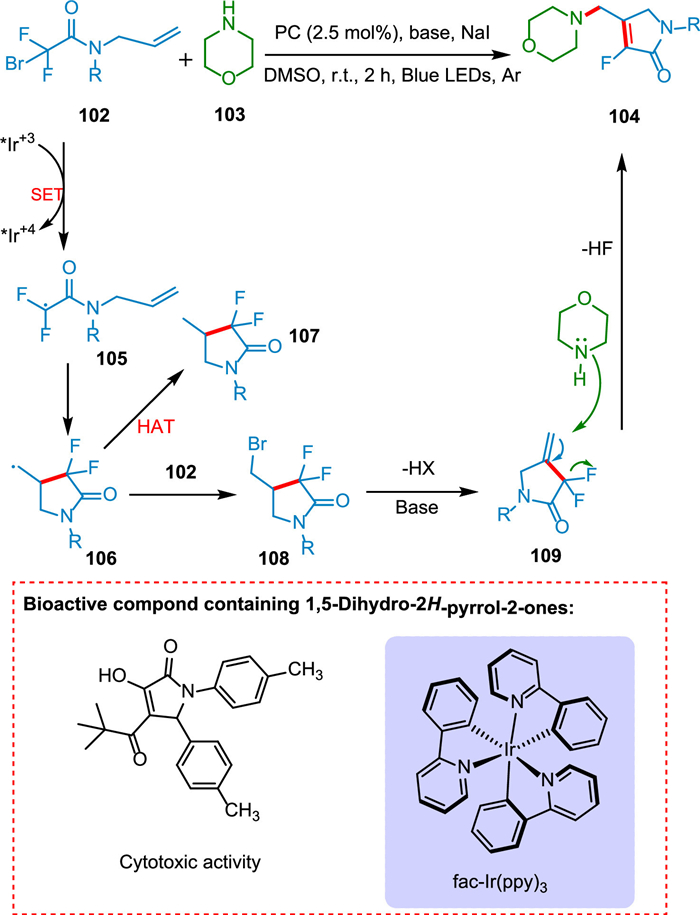
The 1,5-dihydro-2H-pyrrol-2-one is a significant moiety in unsaturated conjugated lactam derivatives present in various pharmaceutics and drugs. Compounds containing 1,5-dihydro-2H-pyrrol-2-one show cytotoxic (antiradical) activity [129]. Ye et al. revealed an unusual photoinduced chain pathway for N-allylbromodifluoroacetamide cyclization/defluorination with cyclic secondary amines. Consequently, a diverse range of valuable 3-fluoro-1,5-dihydro-2H-pyrrol-2-ones were synthesized under mild conditions using easily accessible precursors. A SET from N-allylbromodifluoroacetamide 102 to PC* yields N-allylbromodifluoroacetamide radical 105, which undergoes 5-exo-trig cyclization to generate 106, which reacts with 102 and yields 109 after proton abstraction by a base. The addition of cyclic amine 103 followed by defluorination from 109 yields the targeted product 104 (Scheme 20) [130].

|
Download:
|
| Scheme 20. N-Allylbromodifluoroacetamide cyclization/defluorination with cyclic secondary amines. | |
{kind=link}
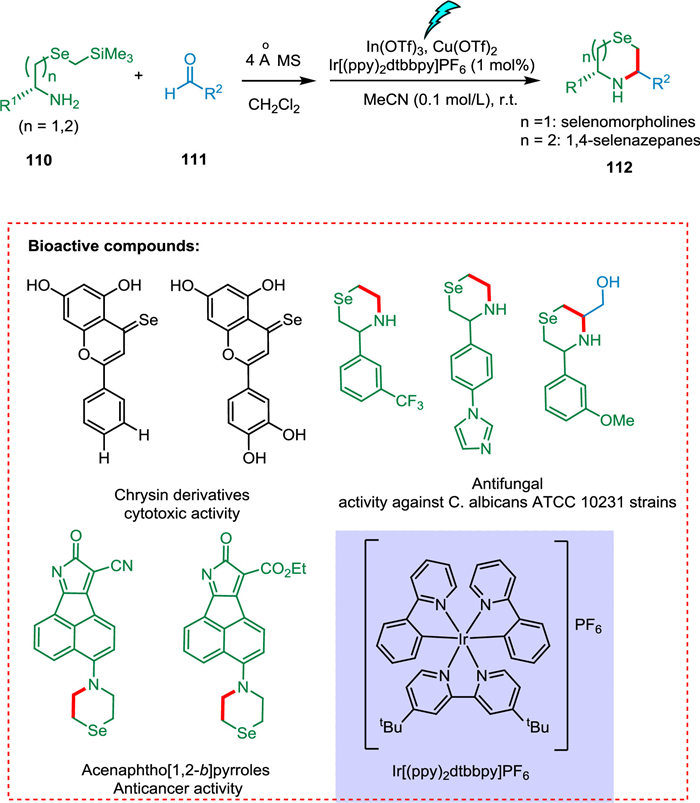
Zhou et al. reported the synthesis of C3/C5-substituted selenomorpholines and 1,4-selenazepanes under mild photocatalytic conditions from various aldehydes by using SLAP reagents containing selenium atoms (Scheme 21). Enantiomerically pure forms as 1,2-amino alcohol and α-amino acid of heterocycles were also prepared. This developed strategy has also modified biologically active molecules in the late stages. Chrysin having selenium embedded in the structure exhibits improved selectivity against cancerous cells compared to its derivatives having O or S. They also exhibit significantly increased anticancer action on nine distinct malignant cell lines. In another case, selenomorpholine derivatives of acenaphtho[1,2-b]pyrroles showed an improved antitumor effect [131].

|
Download:
|
| Scheme 21. The synthesis of C3/C5-substituted selenomorpholines and 1,4-selenazepanes. | |
{kind=link}
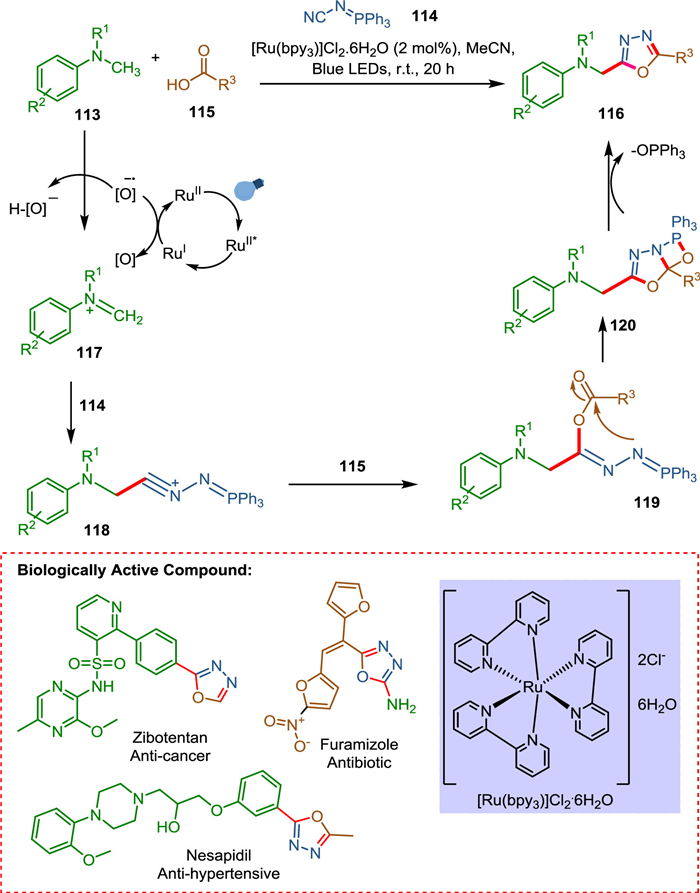
According to Russo et al. 1,3,4-oxadiazole derivative can be synthesized by using a photocatalytic MCR approach utilizing N-alkyl-N-methylanilines, N-isocyanoimino triphenylphosphorane, and carboxylic acids in the presence of visible light. As further evidenced by the late-stage derivatization of pharmaceutical products, this reaction utilizes an extensive substrate range under milder reaction conditions. A two-step one-pot methodology is also published for producing non-symmetrical diacylhydrazines. The N, N-dimethylanilines 113 via an excited ruthenium photocatalyst is converted into the corresponding iminium ion 117, which reacts with isocyanoimino triphenylphosphorane 114 to form a cationic intermediate 118 followed by the reaction with carboxylic acid 115 to form 119 that undergoes intramolecular cyclization resulting in 120. The OPPh3 leaves form 1,3,4-oxadiazole derivatives 116 (Scheme 22). It is worth noticing that the result of the reaction is the formation of three new bonds that is carbon-carbon, carbon-oxygen, and carbon-nitrogen while only losing an equivalent hydrogen atom and one of POPh3, which shows a high atom economy [132]. This novel synthetic method demonstrates the fusion of multicomponent reactions with photoredox catalysis, effectively expanding the chemical space that can be accessed through efficient and cost-effective means [133].

|
Download:
|
| Scheme 22. Synthesis of 1,3,4-oxadiazole derivative by photocatalytic MCR approach. | |
{kind=link}
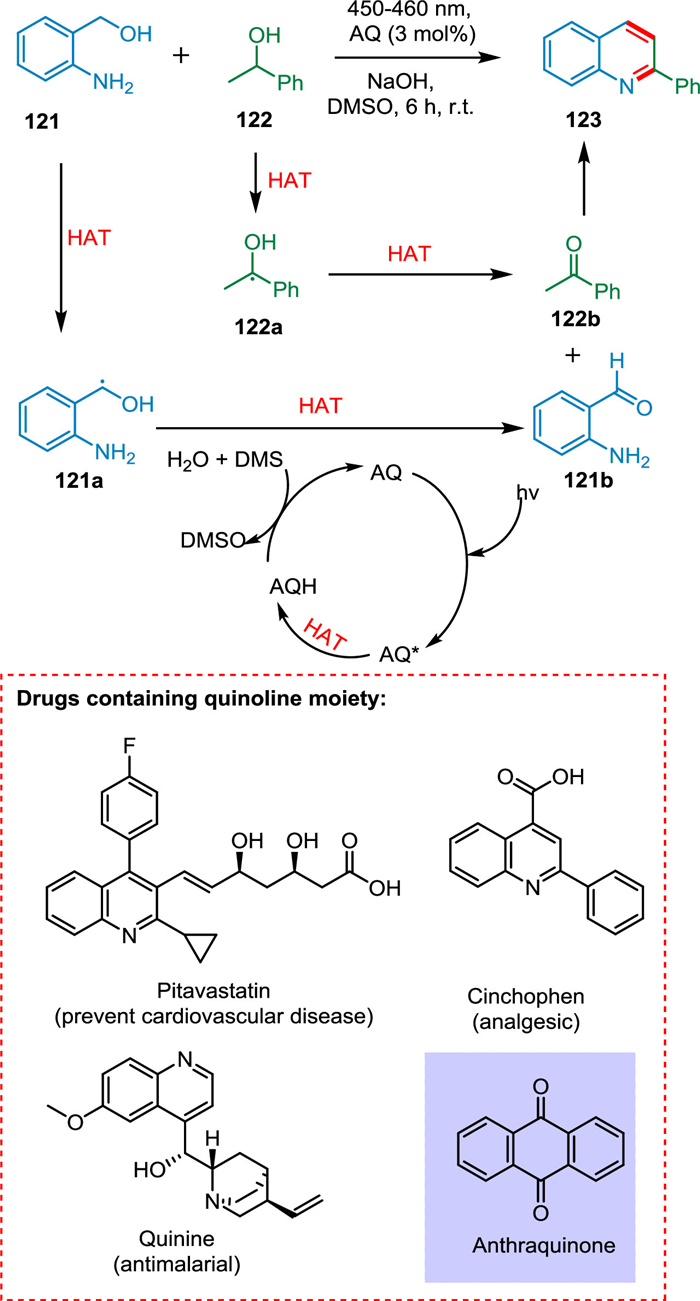
Quinoline has been recognized as an exclusive scaffold due to its promising biological activities proven by approved drugs such as pitavastatin, cinchophen, camptothecin, and quinine [134]. Xu et al. reported the synthesis of quinolines by cyclizing secondary alcohol into 2-aminobenzyl alcohols at room temperature in the presence of light and an organic photocatalyst. This photochemical method eliminated the need for extreme thermal conditions and complex metal-based catalysts by using DMSO and easily accessible AQ as PC. The excited photocatalyst AQ* extracts an H-atom from 121 or 122 via HAT resulting 121a or 122a radicals, followed by another HAT to produce 2-aminobenzaldehyde 121b or acetophenone 122b. The DMSO oxidizes protonated AQ to AQ. Finally, 121b and 122b undergo condensation to form the targeted product 123 (Scheme 23) [135].

|
Download:
|
| Scheme 23. Synthesis of quinolines. | |
{kind=link}
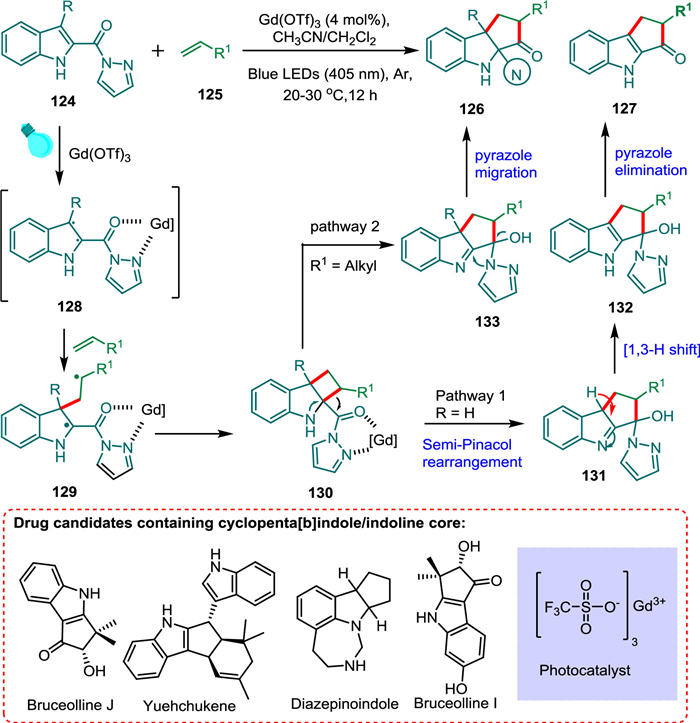
Ma et al. developed a regioselective intermolecular [2 + 2] photocycloaddition/ring expansion that makes use of indoles, Gd3+ photocatalyzed, and may allow divergent approach to both cyclopenta[b]indoles 127 and indolines 126. The easily accessible Gd(OTf)3 salt works best for this visible violet light-mediated transition. The bidentate coordination of 124 to photoexcited PC* forms an excited state 128, which then undergoes [2 + 2] cycloaddition with alkene 125 forming biradical intermediate 129 followed by cyclobutane formation 130 and cyclobutane-expansion cascade [136]. The former 130 expands to a cyclopentane 131. Now, the reaction pathway splits into two. The 1,3-H transfer from 131 and pyrazole elimination from (132) provide the targeted product 127 via route I. The indole substrate 133 is converted into the product 126 via pyrazole migration (route II) (Scheme 24) [24].

|
Download:
|
| Scheme 24. A regioselective intermolecular [2 + 2] photocycloaddition/ring expansion. | |
{kind=link}
4. Reductive cross-coupling
Reductive cross-coupling is simply a new bond formation between two electrophilic centers selectively carbon-carbon bond is formed. That is why also known as electrophile coupling. Metals typically employed are Ni, Pd, Fe, Co, etc., while reductants are Mn, Zn, Mg, etc. [137]. Selectivity in reductive cross-coupling is more challenging than the orthodox cross-coupling method, which utilizes the reactivity aptitude between nucleophiles and electrophiles. Reactivity is favored via a radical pathway [138]. In 2019, Xia et al. used a photocatalytic reductive coupling approach following the radicals combination to form a new C—C bond and synthesize difluorinated 3-pyrazolidinone [139]. In 2022, Tian et al. performed photocatalytic water splitting to use it as an electron donor for reductive coupling in green organic synthesis [140].
Although cinnamic acids have been synthesized using several techniques, each has its complexities, such as reduced efficiency, extended reaction times, decreased catalytic capacity, lower yield, and limited substrate scope. Tripathi and Yadav used visible light photoredox catalysis to create a new, highly stereoselective synthesis of (E)-cinnamic acids 136 from β-nitrostyrene 134 and CBr4 (135). Photoexcited photocatalyst converts 135 into CBr3 radicals 138 and 137. The former 138 is trapped by 134 and generates a stable radical 139, which undergoes denitration forming 140 accompanied by hydrolysis yields 136 (Scheme 25). The (E)-β-nitrostyrenes have broad substrate scope and yield good products, especially when installed in electron-releasing groups. This approach is not favorable for aliphatic nitroalkanes. No product is formed by employing CCl4. Tripathi and Yadav conducted the reaction with eosin Y, but it was not proved as efficient as Ru(bpy)3Cl2 [141].

|
Download:
|
| Scheme 25. Ru(bpy)3Cl2 catalyzed stereoselective synthesis of (E)-cinnamic acids. | |
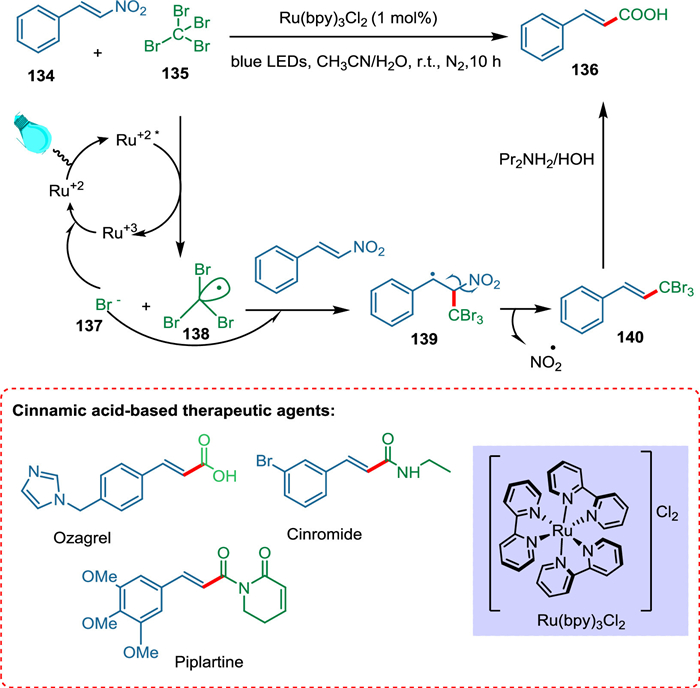{kind=link}
Various strategies have been developed to put acyl groups at the anomeric carbon in glycosides, but those methods use severe conditions and reactive organometallic chemicals. Natural products with glycosidic attachment have improved biological tissue solubility, membrane transport, and specificity [142]. These compounds are abundant and contribute as essential structures to a wide range of anticancer, antibiotic, and type-II antidiabetic drugs [143]. C-Acyl glycosides have been shown to suppress reactive oxygen species, play an essential function in cell signaling, and cause glutamate-induced cell death [144,145]. The photochemical C(sp3)-H acylation of a glycosidic molecule is used to manufacture zaragozic acid C (synthase inhibitor) [19]. Badir et al. used Ni/photoredox dual catalysis to create non-anomeric C acyl glycosides (Scheme 26). The retention of anomeric carbon as a tool for subsequent late-stage functionalization is critical for transition. This process has a wide range of functional group tolerance and is simple to operate. The synthesis of zaragozic acid C is founded on a photochemical C(sp3)-H acylation approach of a glycosidic moiety [146]. A variety of glycosyl-based radicals are produced by an organic photocatalyst and cross-coupled with activated carboxylic acids to produce molecules with potential therapeutic use [147].

|
Download:
|
| Scheme 26. Ni/photoredox dual catalysis to create non-anomeric C-acyl glycosides. | |
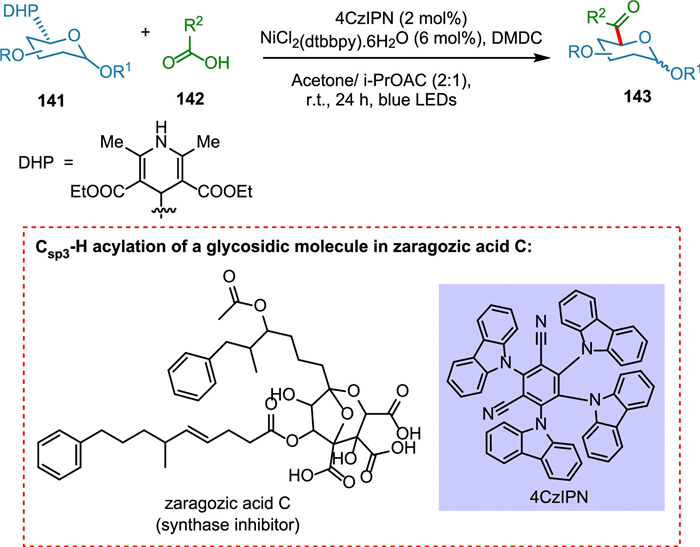{kind=link}
Wang et al. reported trifluoromethylthiolation of aldehydes under redox-neutral conditions using a decatungstate HAT photocatalyst. The procedure is highly selective, easy to perform, and has good functional group tolerance. The TBADT is used as a HAT photocatalyst in this approach to effectively synthesize trifluoromethylthioesters via direct functionalization processes of aldehydic C(O)-H bonds. The HAT occurs from 144 to photoexcited photocatalyst, forming reduced PC and nucleophilic formyl radical 147, which rapidly reacts with the electrophilic reactant 145 to yield the targeted product 146 upon the removal of a Phth-radical 145a, which is reduced to 145b by employing base (Na2HPO4) and reduced PC with concurrent regeneration of ground state PC (Scheme 27). It is useful for late-stage derivitization of pharmaceutical scaffolds, making this a valuable approach in drug synthesis [148].

|
Download:
|
| Scheme 27. Decatungstat catayzed trifluoromethylthiolation of aldehydes under redox-neutral conditions. | |
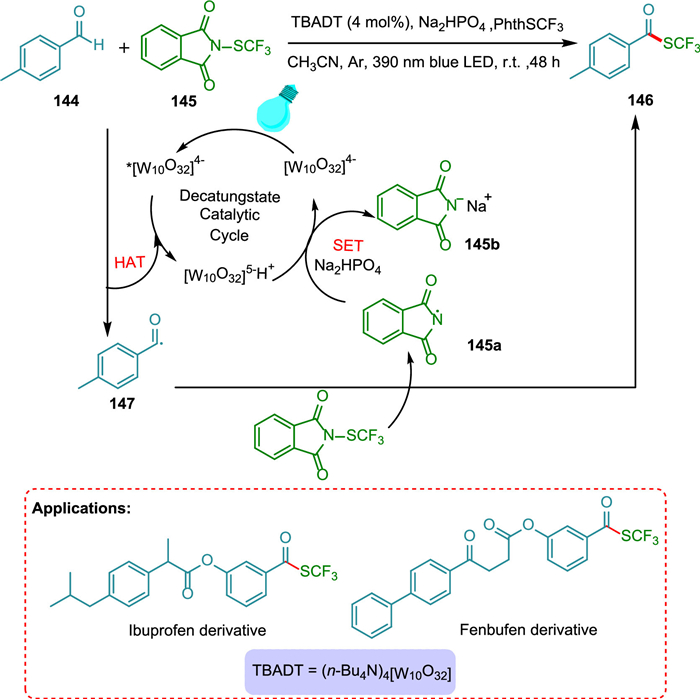{kind=link}
Enantioenriched 1,4-dicarbonyl compounds are structural motifs in ilomastat, batimastat, and (-)-bursehemin and are useful building blocks in pharmaceutical drug production [149]. Kuang et al. developed a moderate approach that uses a combination of chiral rhodium catalyst and neutral eosin Y (HAT photocatalyst) to produce 1,4-dicarbonyl compounds straight from aldehydes efficiently. The photoexcited photocatalyst takes up hydrogen atom from aldehyde 148, forming reduced PC—H and radical of aldehyde 151, which reacts with rhodium-activated 1,4-dicarbonyl derivative 152 creating radical intermediate 153. The HAT from reduced PC—H to 153 results in the formation of rhodium coordinated compound 154 followed by the removal of the rhodium catalyst yielding the targeted product 150 (Scheme 28). This process stands out for its ease of operation, atom economy, high yields (up to 99%), as well as the enantioselectivity (up to 99% ee) of the produced products [150].

|
Download:
|
| Scheme 28. Dual catalysis approach synthesizing 1,4-dicarbonyl compounds straight from aldehydes. | |
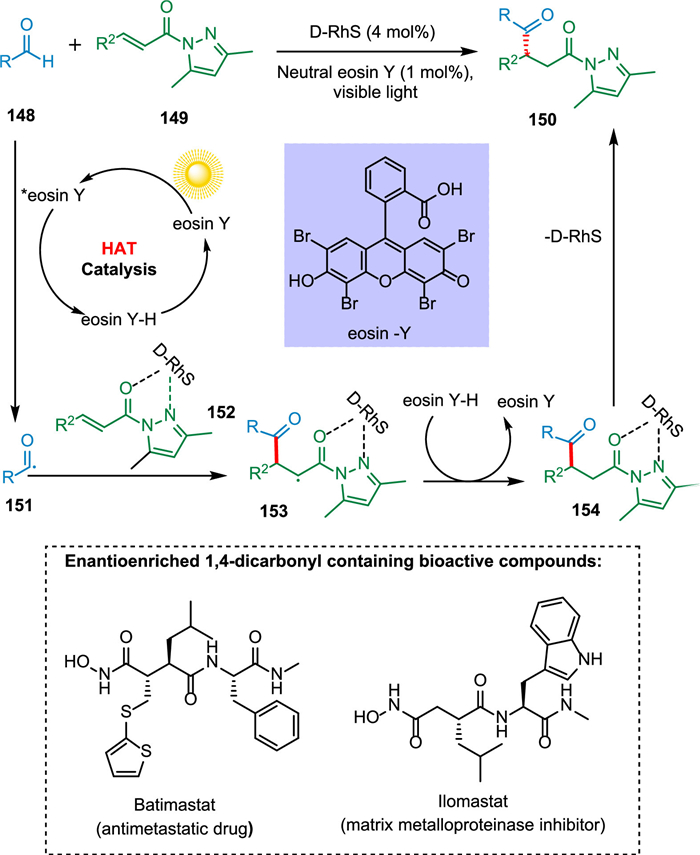{kind=link}
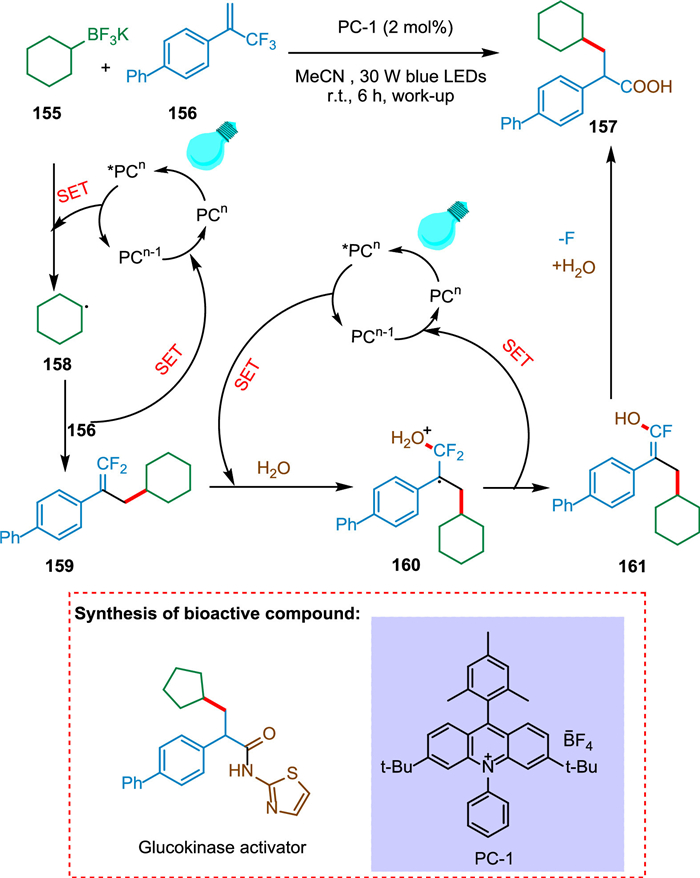
Li et al. proposed that at ambient temperature, a photocatalyzed multicomponent triple defluorinative functionalization of α-trifluoromethyl alkenes yields an array of α-arylated carboxylic acids, primary, secondary, and tertiary amides [151]. The photoexcited photocatalyst *PCn undergoes SET with 155 producing radical intermediate 158 and PCn-1 [152,153]. The former 158 combines with radical species 156 to form benzylic radical 160. The first catalytic cycle completed by SET from PCn-1 to 160, yields gem‑difluoroalkenes 161 (Scheme 29) [154]. The third C-F bond is cleaved by the second β-fluoride elimination and acyl substitution with oxygen nucleophile [151]. The PC-1 and H2O were needed to carry out this reaction, and the yield was lowered as the work-up was not done. The reaction involves three separate C-F bond cleavages, allowing the conversion of methyltrifluorides into synthetically useful functional groups. The trifluoromethyl alkenes are coupled with alkyltrifluoroborates, water, and amines to produce α-arylated carboxylic acids and amides. As a result, these key motifs can now be constructed from basic and readily available starting materials [151].

|
Download:
|
| Scheme 29. Photocatalyzed multicomponent triple defluorinative functionalization of α-trifluoromethyl alkenes. | |
{kind=link}
5. C-H activation
LSF has its mark in drug and natural product synthesis exploring new drug analogous. In late-stage functionalization (LSF), both participating reactants' radicals combine forming new bonds like carbon–carbon, carbon–nitrogen, carbon–oxygen, etc. One moiety having a C—H bond undergoes homolytic cleavage via SET/HAT generating carbon radical [155]. Sustainability [156], high step-economy [157], no need for prefunctionalization, and new disconnection approach [158] make C—H functionalization even more significant. In 2019, Vidyacharan et al. synthesized a liver X-receptor inhibitor drug by C—H functionalizing indazoles with an aryl group at C-2, employing eosin Y as a photocatalyst [159]. In 2021, Hou et al. formulated a metal and oxidant-free photocatalytic approach to acylate pyridine N-oxide at C-2 and applied this strategy to acrivastine synthesis, an important drug intermediate [160]. In 2023, external photocatalyst-free C—H activation has been reported [161,162].
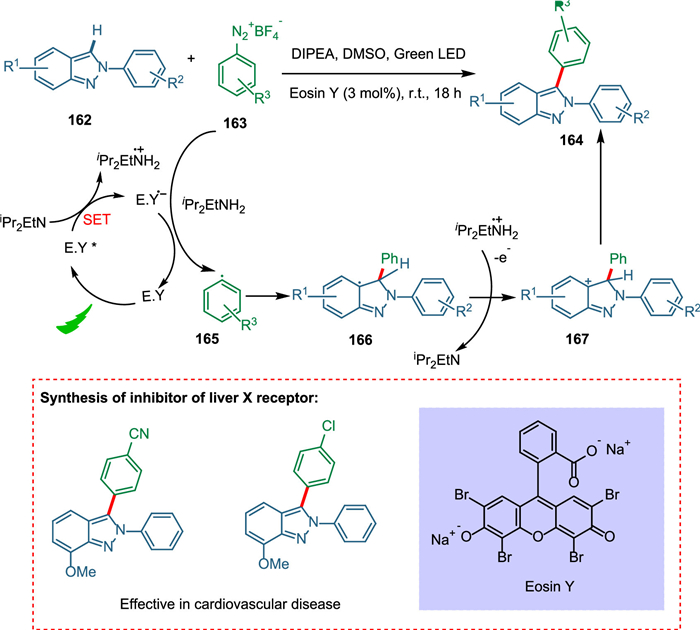
In the pharmaceutical industry, indazoles offer a broad spectrum of bioactivity [163], including antimicrobial [164], anti-inflammatory [165], HIV protease inhibition [166], and anticancer agents [167]. Indazole is a basic scaffold in commercially relevant medicinal products such as MK-4827 and pazopanib [168]. Most of the cases of Negishi cross-coupling [169] and arylation by aryl halides [170,171], which needed Cu/Pd as catalysts, costly ligands, and use of base and oxidant as additives having prolonged reaction times. So, less work is reported for direct arylation of 2H-indazoles at the C-3 position. Vidyacharan et al. proposed a simple eco-friendly methodology for direct C-3 arylation of 2H-indazoles with aryldiazonium salts using organo-photoredox catalysis with green light under milder conditions and synthesized inhibitor drugs of liver X-receptor without converting DIPEA into its radical cation. The SET from reduced PC completes the catalytic cycle forming aryl radical 165 and recovering the photocatalyst too. The resultant radical moiety 165 reacts with heteroarene 162, giving radical intermediate 166, which is changed to carbocation intermediate 167 by supplying electrons to the DIPEA. The deprotonation aromatizes the system and yields the targeted product 164 (Scheme 30) [159]. The phenyl moiety with electron-donating groups resulted in high yields, whereas bromosubstitution yielded extremely low yields. Unfortunately, N-2 substituents of 2H-indazoles did not give targeted products due to decreased aromatic nature of the parent ring [172].

|
Download:
|
| Scheme 30. Organo-photoredox catalyzed direct C-3 arylation of 2H-indazoles. | |
{kind=link}
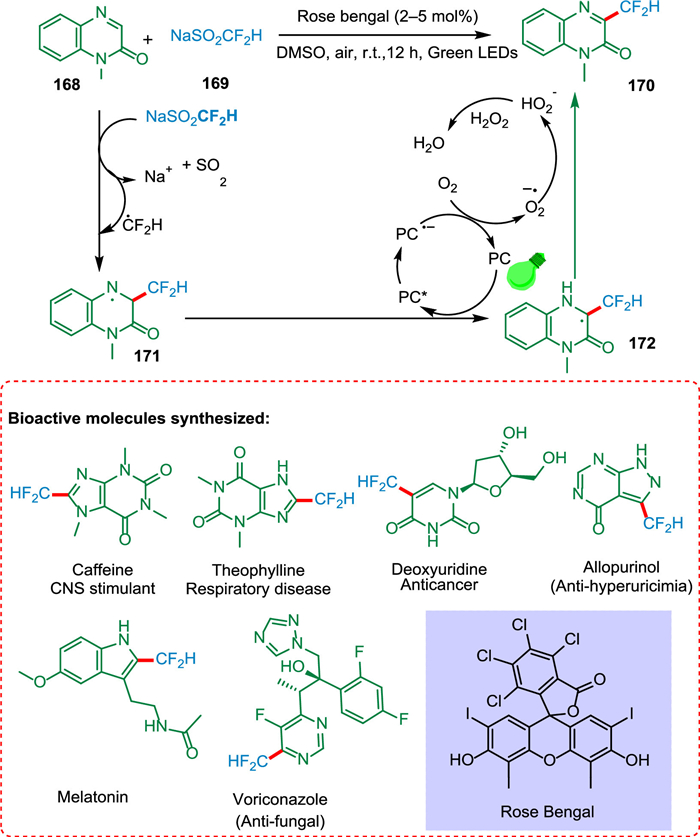
Zhang et al. devised a light-driven direct C—H difluoromethylation of heterocycles by employing NaSO2CF2H as a source of CF2H radicals. The green oxidizing agent O2 is used in this process with no metal additives and mild conditions. The difluoromethylation of carbon-hydrogen bond transforms nitrogen-containing heterocyclic molecules 168 into N-radical species 171. The C-radical specie 172 is the product of intramolecular radical transfer. The N—H homolytic cleavage produces the desired product 170 (Scheme 31). Furthermore, certain nitrogen-containing bioactive compounds can be directly difluoromethylated via this technique. Additionally, the difluoromethylation approach might help drug discovery because 2′-deoxy-5-difluoromethyluridine (F2TDR) has potential action against various cancer cell lines [173].

|
Download:
|
| Scheme 31. Light-driven direct C—H difluoromethylation of heterocycles. | |
{kind=link}
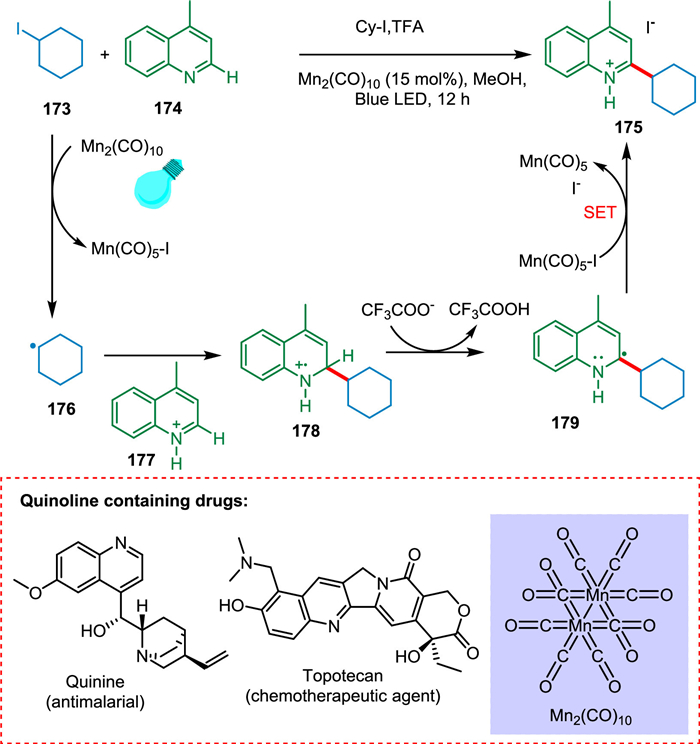
Nuhant et al. established a photoredox Minisci reaction with unactivated iodoalkanes in the presence of Mn complex, Mn2(CO)10 as a catalyst. This mild reaction is light-mediated and has good functional group tolerance. Photocatalyst converts iodocyclohexane 173 to cyclohexyl radical 176. The protonated 4-methylquinoline 177 reacts with 176 to yield a cation radical intermediate 178 followed by HAT, resulting in 2-cyclohexyl-4-methyl-1,2-dihydroquinoline 179. The SET results in 175 (Scheme 32). Nuhant et al. showed this approach with remarkable success for late-stage functionalization of complicated medicines [174].

|
Download:
|
| Scheme 32. Photoredox Minisci reaction with unactivated iodoalkanes. | |
{kind=link}
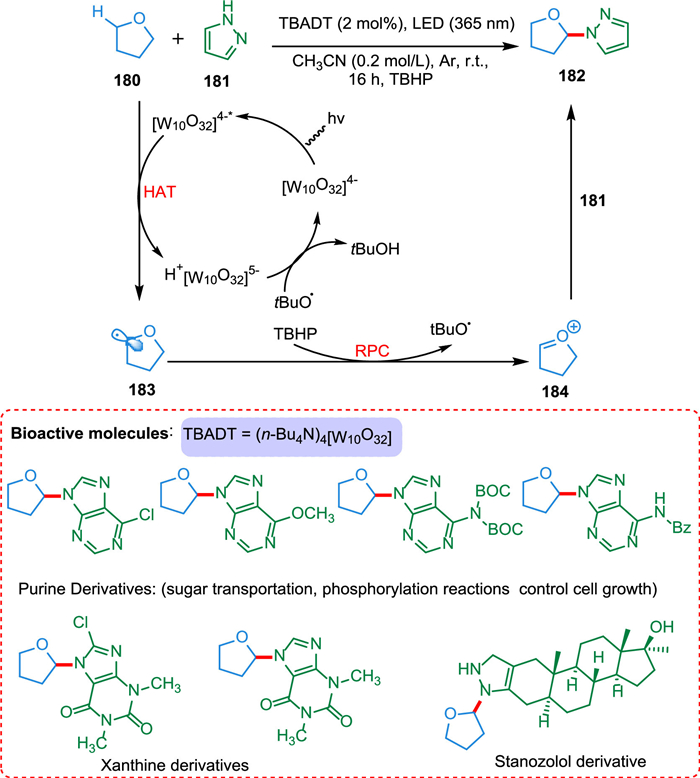
Photocatalytic HAT is a highly effective strategy for C(sp3)-H functionalization of organic compounds. Wan et al. established decatungstate HAT photocatalysis for C(sp3)-H oxidative heteroarylation using the radical polar cross-over (RPC) approach. The current strategy is scalable and has broad substrate scope. There is no need for prefunctionalization [175]. The breaking of the C—H bond in α-position regarding heteroatom 180 by PC* gives a carbon-centered radical 183, which gets oxidized by TBHP, yielding a stable electrophilic oxocarbenium ion 184 which, on reaction with 181, forms carbon–nitrogen bond 182 (Scheme 33). The [W10O32]4−, which is HAT photocatalyst has selectivity, robustness, and is easy to synthesize [176]. This methodology can be applied for the late-stage modification of bioactive products, that is stanozolol, (-)-ambroxide, podophyllotoxin, and dideoxyribose [175].

|
Download:
|
| Scheme 33. Decatungstate catalyzed C(sp3)-H oxidative heteroarylation. | |
{kind=link}
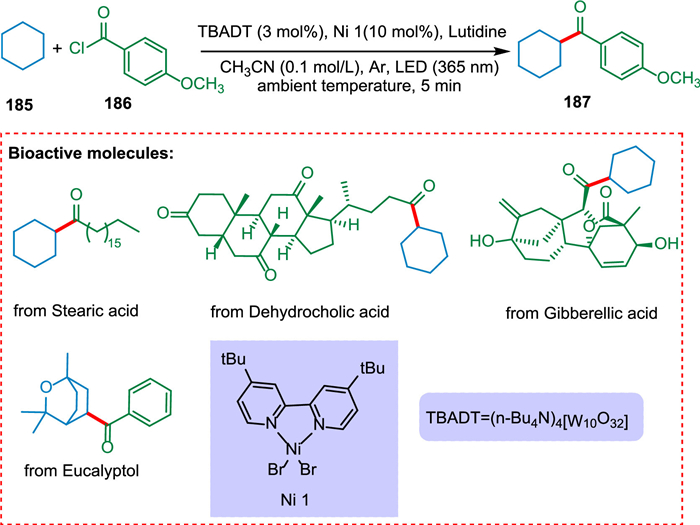
Mazzarella et al. described a photocatalytic process for the acylation/arylation of unfunctionalized alkyl derivatives. The [W10O32]4− creates carbon-centered nucleophilic radicals. The Ni catalyst captures these radicals and eventually forms C(sp3)-C(sp2) bonds (Scheme 34). Mazzarella et al. also functionalized complicated organic or natural scaffolds at a late stage [177].

|
Download:
|
| Scheme 34. Acylation/arylation of unfunctionalized alkyl derivatives. | |
{kind=link}
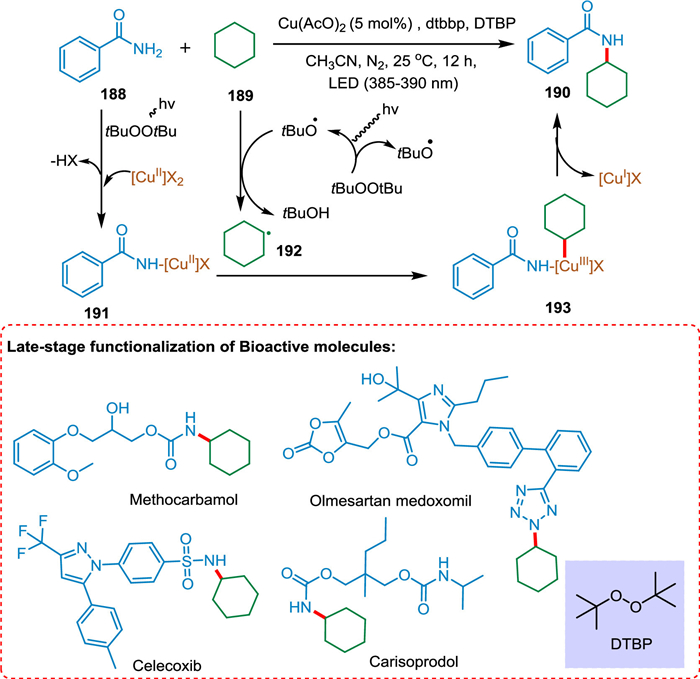
Zheng et al. established a light-mediated copperII-peroxide method for alkylating amides and nitrogen nucleophiles with unactivated alkanes. This strategy can be employed for the late-stage N-alkylation of bioactive compounds. Under the action of peroxide and light, the [CuII]-amide complex 191 is produced from a copper source and the amide 188. The photolysis of DTBP forms the tBuO radical. The HAT from cyclohexane 189 to tBuO radical creates cyclohexyl radical 192, which gets coordinated with the [CuII]-amide complex 191, resulting in the formation of the [CuIII] complex 193. The coupling product 190 is formed via reductive elimination from 193. The [CuII]-phth complex is a crucial component (Scheme 35) [178].

|
Download:
|
| Scheme 35. Alkylation of amides and nitrogen nucleophiles with unactivated alkanes. | |
{kind=link}
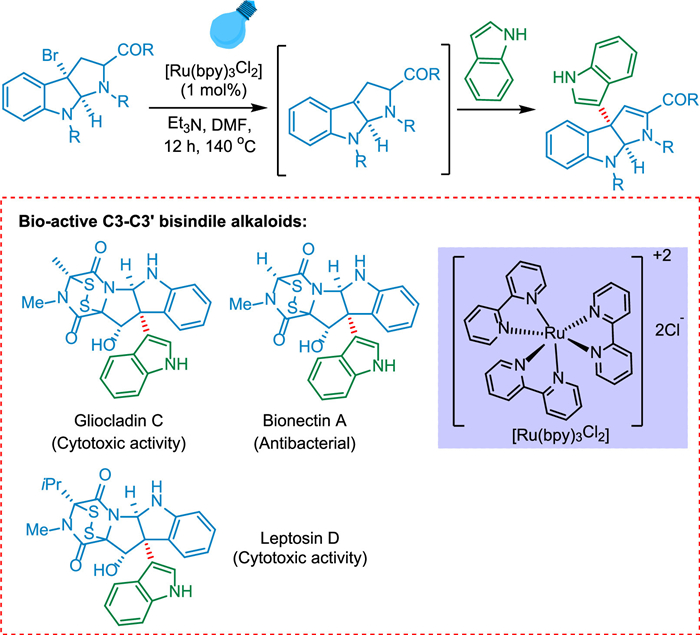
The C3-C3′ indole, including the 3a-(3-indolyl)-hexahydropyrrolo-[2,3-b]indole skeleton that is drugs like gliocladine C [179], leptosin D [180], and bionectins [181], which are antibacterial and cytotoxic, respectively. Furst et al. prepared 30% of gliocladin C in 10 stages using Boc-D-tryptophan methyl ester that was easily accessible in the market (Scheme 36). This study emphasizes photoredox catalysis as an effective technique for this specific synthesis and a reliable way to obtain a range of complicated compounds [182].

|
Download:
|
| Scheme 36. Synthesis of gliocladin C. | |
{kind=link}
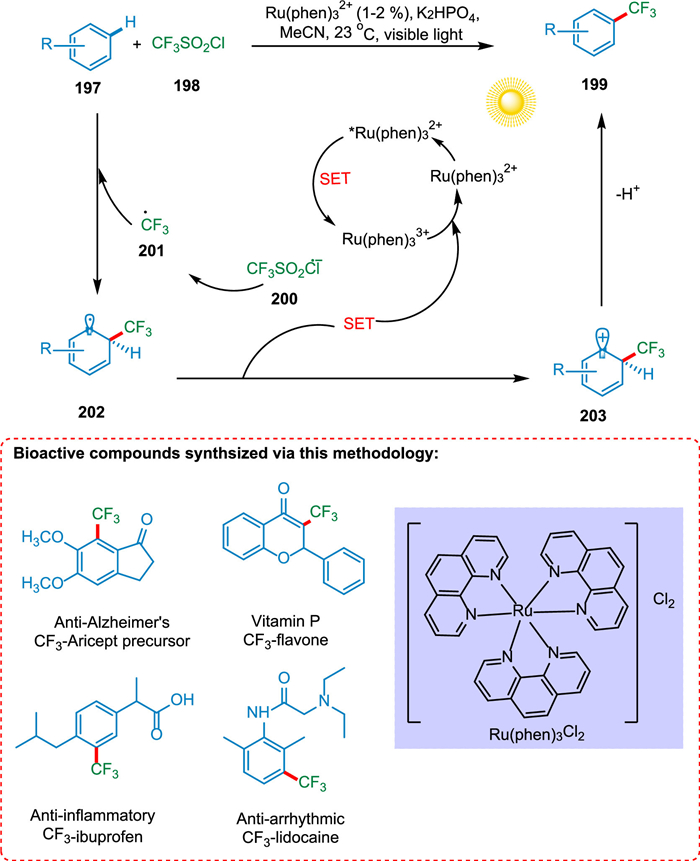
Nagib and MacMillan's photoredox-based method is free from prefunctionalization and allows trifluoromethylation of aromatic and heteroaromatic materials. The significance of this conversion has been emphasized by the trifluoromethylation of bioactive compounds using affordable PC and an ordinary light source. The SET from photoexcited PC* *Ru(phen)32+ to triflyl chloride 198 creates CF3 radical 201 and Ru(phen)33+. The CF3 radical 201 is formed by splitting the CF3SO2Cl radical anion 200. The SET from cyclohexadienyl radical 202, formed by the addition of the CF3 radical 201 to benzene 197, to oxidized PC (RuIII) forming a cationic intermediate 203, and PC is regenerated. In the end, the arene prefunctionalization is not required because the cyclohexadienyl cation 203 may be deprotonated with a base to produce the required trifluoromethyl arene 199 (Scheme 37) [183].

|
Download:
|
| Scheme 37. Trifluoromethylation of aromatic and heteroaromatic compounds. | |
{kind=link}
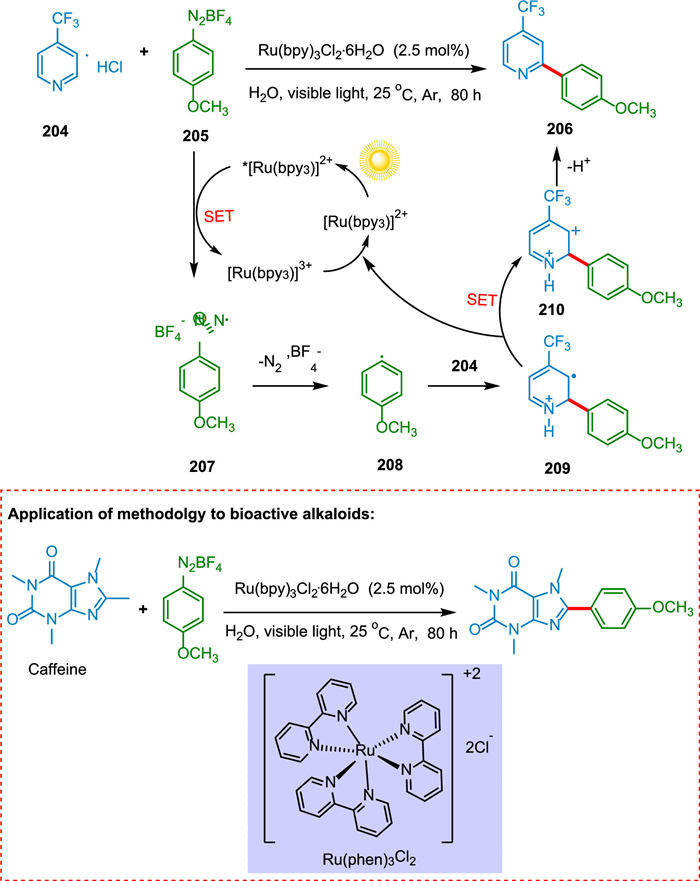
Baran proposed a "radical-type" arylation of N-heterocycles using boronic acids [184]. On the other hand, the Gomberg-Bachmann reaction is the coupling of arene compounds with diazonium salts using base-forming biaryls. This reaction has a broad substrate scope yet low efficiency due to the numerous side reactions [185]. Xue et al. proposed "radical type" coupling of N-heteroarenes with aryldiazonium ions in the presence of light and water as solvent. The SET from photoexcited *[Ru(bpy)3]2+ to the aryldiazonium salt 205 creates phenyl radical 208 and the oxidized [Ru(bpy)3]3+. The phenyl radical 208 is added to the pyridine hydrochloride 204 to creat radical 209, which is then converted to cationic intermediate 210 followed by deprotonation to form coupling adduct 206 (Scheme 38). This reaction has a broad substrate scope of pyridine, and only monosubstituted products with varying regioselectivities are produced. Various xanthenes, thiazole, pyrazine, and pyridazine are appropriate in this approach when aqueous formic acid is used as the solvent. This process is a feasible and environmentally acceptable due to water as a solvent, broad substrate scope, and mild reaction conditions to prepare the compounds bearing aryl-heteroaryl motifs [186].

|
Download:
|
| Scheme 38. The "radical type" coupling of N-heteroarenes with aryldiazonium ions. | |
{kind=link}
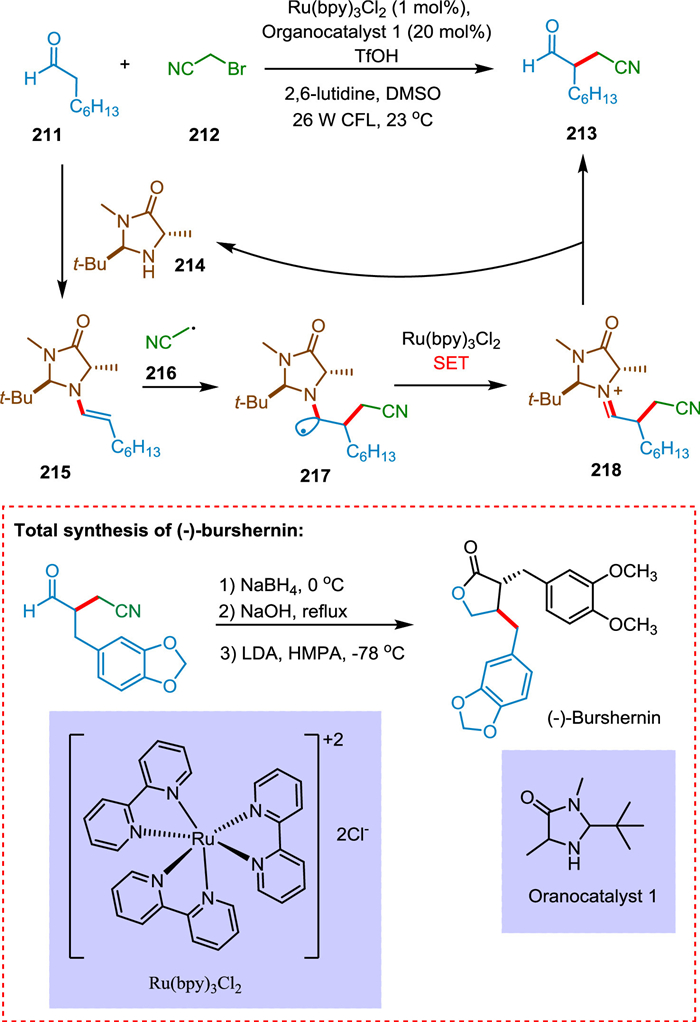
Welin et al. reported that by combining photoredox and enamine catalysis, preparation of enantioselective α-cyanoalkylation of aldehydes is possible. This synergistic catalysis method enables the diversification of oxonitrile products into various drug derivatives and heterocycles. The bromonitrile species 212 is reduced by the highly reducing Ru(bpy)3+ ion via SET yielding cyanoalkyl radical 216. Concurrently, an amine catalyst 214 condenses with an aldehyde 211, forming an enamine having chirality 215, which couples with an electron-deficient radical 216 to create an amino radical 217. The organocatalytic cycle is completed when photoexcitation of Ru(bpy)32+ causes SET to produce a-amino ion 218, which upon hydrolysis produces the α-cyanoalkylated aldehyde 213 simultaneously recovering amine 214 (Scheme 39). This approach has also accomplished the natural lignan product (-)-bursehernin's complete synthesis [187].

|
Download:
|
| Scheme 39. Synthesis of enantioselective α-cyanoalkylation of aldehydes. | |
{kind=link}
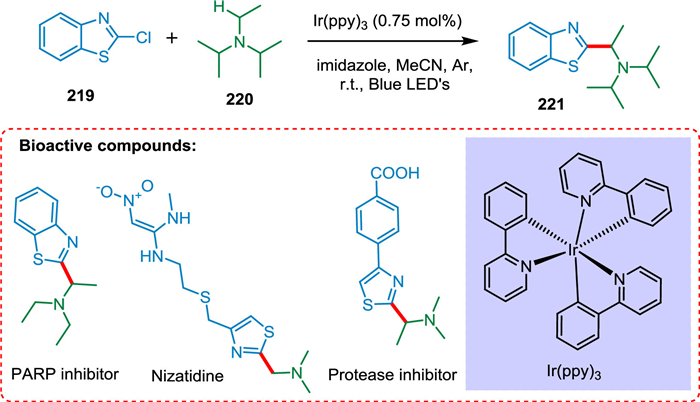
Singh et al. explained the conditions for tertiary aliphatic amines' C—H functionalization following their coupling with various 2-chloroazole derivatives. The photocatalyst tris-fac-Ir(ppy)3 is aided by blue light irradiation and proceeds in a mild and practical environment (Scheme 40). The majority of couplings occur with great regioselectivity. The procedure permits quick access to R-azole carbinamines, frequently present in post-translationally modified peptides, and has excellent functional group tolerance [188].

|
Download:
|
| Scheme 40. C—H functionalization of tertiary aliphatic amines. | |
{kind=link}
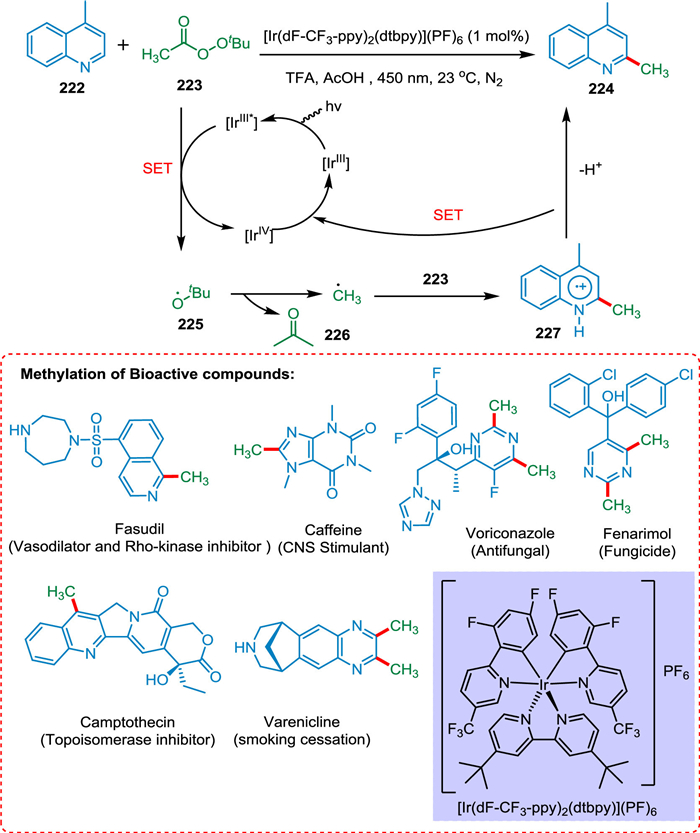
Previous approaches require powerful oxidants and high temperatures to form the required radical species, so small alkyl groups are not inserted in complex formation. DiRocco et al. proposed that by employing a photocatalyst and light-mediated approach, stable peroxides are formed, which are used in the alkylation of an array of heterocycles present in bioactive compounds. The tBPA 223 via PCET yields tBuO radical 225, which is converted into methyl radical 226 via the β-scission. The addition of methyl radical 226 to the protonated heterocycle 222 yields radical cation intermediate 227 followed by HAT to the oxidized PC [IrIV], resulting in the targeted product 224 (Scheme 41). This approach is a significant tool for drug discovery due to its straightforward methodology, mild response conditions, and unusual tolerability [189].

|
Download:
|
| Scheme 41. Photocatalyzed alkylation of heterocycles. | |
{kind=link}
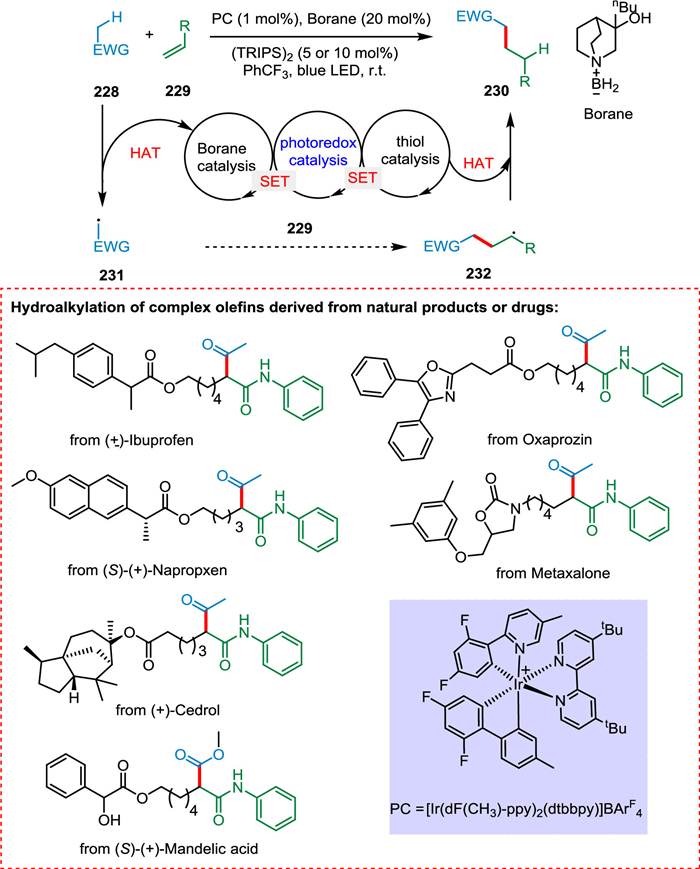
Without prefunctionalization, hydroalkylation of alkene mediated by HAT provides a simple way to produce C(sp3)-rich compounds. In contrast, Giese-type hydroalkylation of unactivated olefins is still tricky due to a lack of ideas for dealing with the associated polarity mismatch. Lei et al. used dual HAT catalysis to accomplish this objective in the presence of light, using catalytic quantities of an amine-borane and thiol as H-atom abstractor and donor, respectively. A substrate with electrophilic C—H bonds 228 undergoes HAT via borane catalysis to yield radical specie 231, which is alkylated with an unactivated alkene 229 to form free radical species 232. The resultant radical intermediate abstracts hydrogen resulting in the product 230 via thiol catalysis (Scheme 42). The reaction has a high atom economy and good substrate scope [190].

|
Download:
|
| Scheme 42. Dual HAT catalysis of hydroalkylation of alkene. | |
{kind=link}
The HAT is a fundamental stage in free-radical processes [191,192]. Advancements in photocatalyzed reaction revolutionized [193,194] light-mediated HAT as an efficient technique for creating molecular complexity from easily accessible starting materials without requiring prefunctionalization [191]. The polarity-matching effect makes compound functionalization via HAT restricted to substrates having hydridic RH (R = C, Si, B, etc.) bonds [195]. The nucleus-loving radicals formed via HAT reagents must exist to render electrophilic carbon-hydrogen bonds suitable to remove hydrogen atoms.
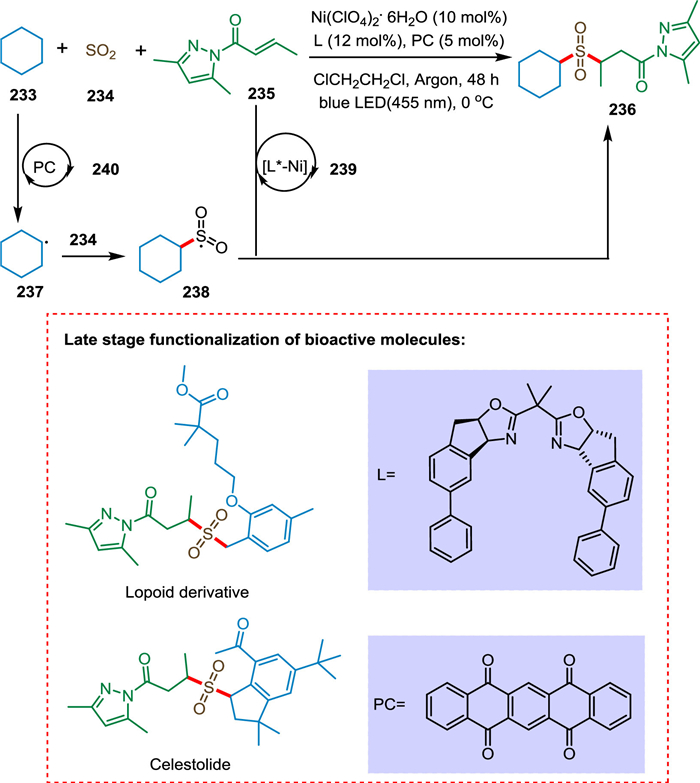
Direct and selective C(sp3)-H functionalization is an emerging approach due to the inexpensive starting materials and excellent atom economy. Cao et al. devised a three-component (C(sp3)-H precursor, SO2 substitute, and α, β-unsaturated carbonyl molecule) asymmetric sulfonylation reaction enabling direct functionalization under mild conditions to yield an array of medically relevant α-C chiral sulfones. The cycloalkane 233 is converted into cycloalkane radical 237 via the photocatalytic cycle 240, which gets trapped by SO2 234 to generate the stabilized sulfonyl radical 238. The compound 235 interacts with the metal-coordinated Michael acceptor 239 due to electronic and steric phenomena. Finally, the ligand exchange results in the chiral sulfone product 236 (Scheme 43). This approach is suitable for the late-stage functionalization of bioactive compounds opening a novel route to enantioenriched drugs derived from hydrocarbon compounds [196].

|
Download:
|
| Scheme 43. Asymmetric sulfonylation reaction enabling direct functionalization. | |
{kind=link}
6. Ring opening
Ring-opening is a stability dependant process. Strained rings tend to open more easily like a five-membered ring undergoes ring opening rapidly compared to a six-membered ring. The smaller the ring size, the more strained it will be and the more easily it will undergo ring opening reactions [197]. In 2019, Zhang et al. reported oxidant-free ring opening of cyclobutanone oxime esters in visible light and PC to synthesize alkylnitrile derivatives of various heterocycles [198]. In 2022, Zhu et al. reported ring opening of sulfonium salts by using hetero-arene moieties in the presence of light and photocatalyst Cu-complex [199].
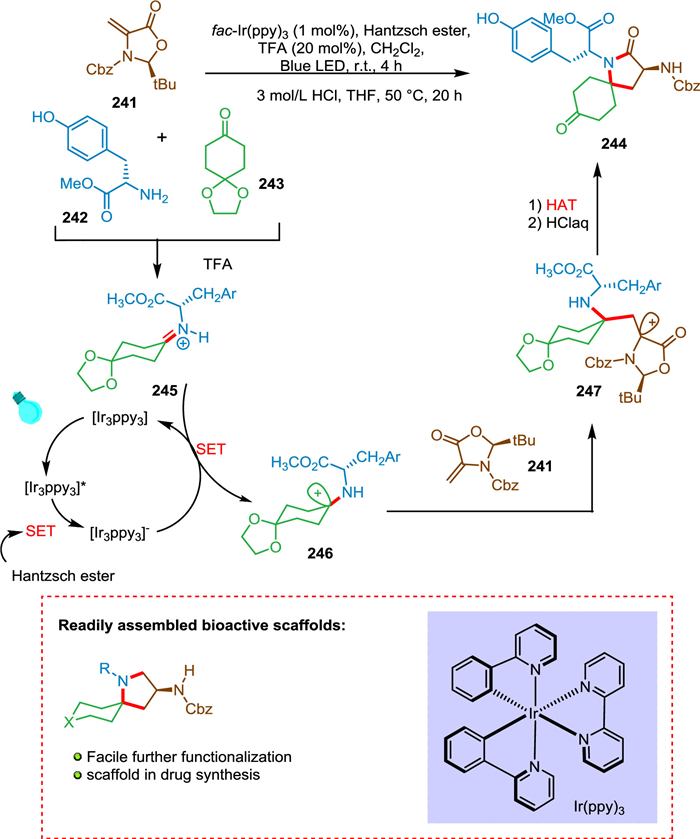
Reich et al. described a multicomponent photocatalytic hydroaminoalkylation method for producing the azaspirocycle with amino substitution on the ring, which is present in (-)-FR901483 and (+)-TAN1251C. This approach is key to synthesize spirolactam moiety via a novel synthesis strategy. This technology finds use in natural products since starting materials are readily available and it is simple to operate. Spirolactam was synthesized by the photocatalytic olefin hydroaminoalkylation, followed by further three steps to produce the TAN125C equivalent, resulting in a high yield (Scheme 44). The FR901483 has an immunosuppressive effect that is extraordinarily potent (more than 100 times stronger than cyclosporine) [45].

|
Download:
|
| Scheme 44. Hydroaminoalkylation method for producing the azaspirocycle with amino substitution on the ring. | |
{kind=link}
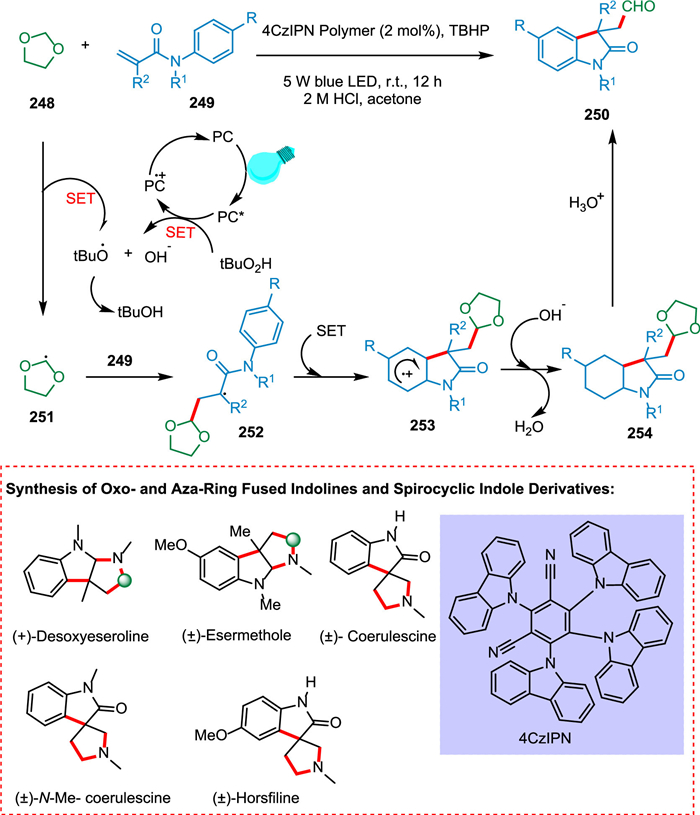
Ou et al. designed a fast production of 3-formyloxindoles by double C—H functionalization of N-arylacrylamides and 1,3-dioxolane in a metal-free and light-mediated manner employing a heterogeneous photocatalyst [200]. The photoexcited PC* undergoes SET to TBHP and yields a tert‑butyloxy radical. The regioselectivity of tert‑butyloxy radical then abstracts a proton from C-2 of 1,3-dioxolane 248 to create radical specie 251 [201], which adds to acrylamide 249 to form radical specie 252 undergoing intramolecular cyclization to yield radical intermediate 253. The dioxolane acetal intermediate 254 is produced by oxidizing radical intermediate 253 with the PC•+, subsequently regaining aromaticity. Ultimately, the acidic hydrolysis yields the required product 250 (Scheme 45). This study illustrates the tremendous promise of employing CMPs, as light-triggered photocatalysts with stability, reusability, and metal-free characteristics that offer sustainable production of bioactive heterocycles and medicinal molecules. The obtained 3-formyloxindoles act as an essential component for the quick generation of an array of pharmaceutically essential products such as (±)-desoxyeseroline, (±)-esermethole, and (±)-N-Me-coerulescine, in better amounts in comparison to multistep reactions [200].

|
Download:
|
| Scheme 45. Synthesis of 3-formyloxindoles by double C—H functionalization of N-arylacrylamides and 1,3-dioxolane. | |
{kind=link}
7. Oxidative cross-coupling
In synthetic chemistry, selective C—C bond or C-heteroatom bond formation is tricky. In this regard, cross-coupling reactions contribute significantly. Heck, Suzuki, and Negishi won the Nobel Prize in 2010 for palladium-catalyzed cross-coupling reaction. Conventional cross-coupling reaction operates between nucleophilic and electrophilic reactants and has some limitations as lack of sustainability, requiring prefunctionalization and side products (low step economy). Oxidative coupling involves the release of two electrons from both nucleophilic reactants with the use of various oxidants and catalysts. Oxidative coupling resolves the above limitations of conventional cross-coupling reaction as its nucleophilic center reactants have hydrogen atoms attached to carbon or heteroatoms. Thus, prefunctionalization and oxidants are unnecessary too [202]. Song et al. proposed the metal-free, semi-heterogeneous g-C3N4/iodide salt dual catalysis potent for forming oxidative bonds [203].
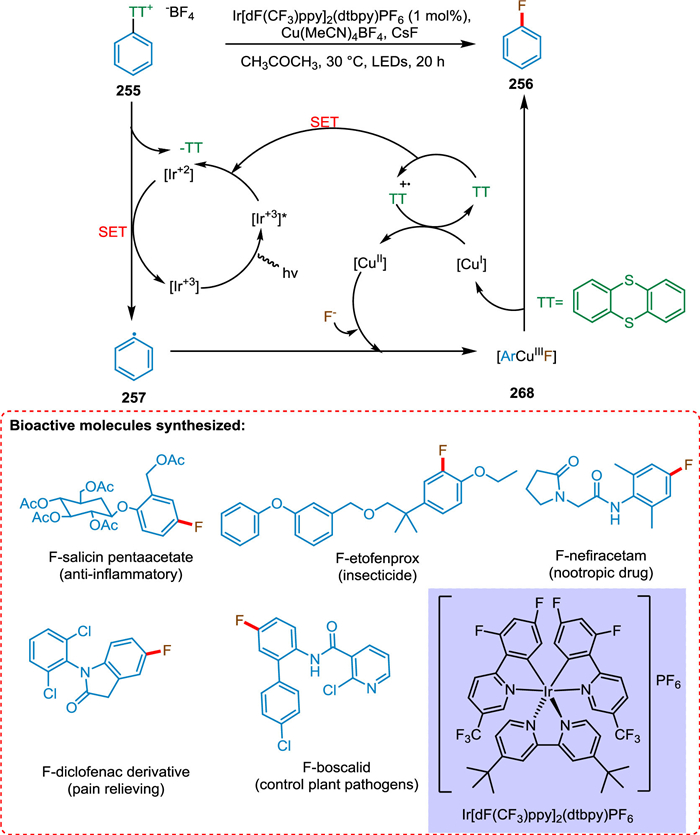
According to Li et al., photoredox catalysis helps make bonds that are difficult to form in conventional redox catalysis, particularly when transition metals are used as PC. The ArTT+ salt 255 via SET from reduced iridium PC goes converted into aryl radical 257, which coordinates with Cu redox catalyst to form a copper-aryl coordinated intermediate 258. The Cu is removed in reduced form, yielding a fluorinated aryl compound 256 (Scheme 46). The leaving groups useful in traditional cross-coupling catalysis, like bromide, are being utilized effectively in metallophotoredox catalysis for arene functionalization. However, most aryl bromides are not reduced by usual photoredox catalysts, making it difficult to produce synthetically valuable aryl radicals directly. So, the redox potential of leaving group and photocatalyst should match for better reactivity and selectivity. That is complicated coupling reactions by using arylcopper-III complexes. Li et al. demonstrated the prospective significance of aryl thianthrenium salts and the application of this photoredox metal PC in late-stage aromatic fluorination [204].

|
Download:
|
| Scheme 46. Fluorinaton of aryl thianthrenium salts. | |
{kind=link}
8. Dehydrogenation
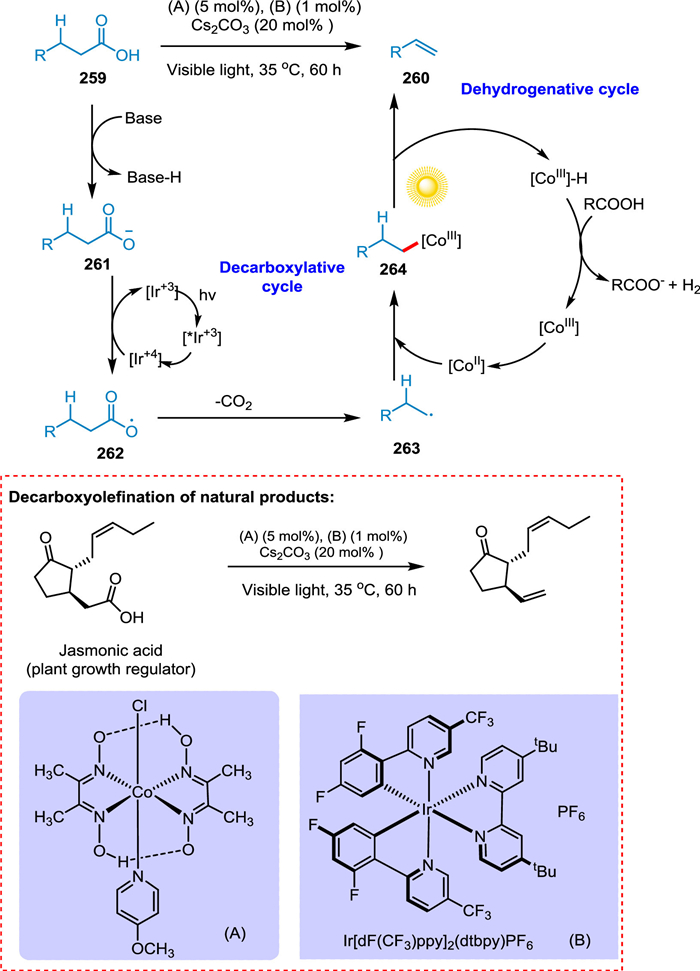
Sun et al. described a decarboxyolefination method that does not require stoichiometric additions, paving the way for developing an approach for the green synthesis of alpha-olefins from renewable carboxylic acids. The carboxylic acid 259 is converted by base into carboxylate anion 261, which then undergoes a decarboxylative cycle with the aid of a PC to create alkyl radical 263, which then undergoes a dehydrogenative cycle to yield 260 (Scheme 47). This approach can be applied in the late-stage functionalization of natural products and even in drug discovery [205].

|
Download:
|
| Scheme 47. A decarboxyolefination method synthesizing alpha-olefins. | |
{kind=link}
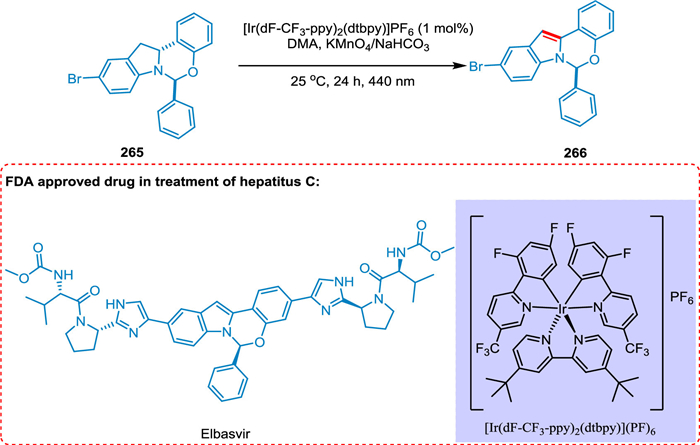
A potent NS5A antagonist used to treat chronic hepatitis C is elbasvir. Yayla et al. reported the identification and investigation of indoline dehydrogenation for the photoredox-mediated synthesis of elbasvir. The Ir[dF(CF3)ppy]2(dtbbpy)PF6 and the ecologically safe oxidant tert‑butyl peroxybenzoate have been proven as good photoredox catalyst and oxidant combination yielding the targeted product with excellent yields without disturbing optical purity through multi-parallel experiments (Scheme 48). A pharmaceutical intermediate is produced for the first time using photoredox catalysis [206].

|
Download:
|
| Scheme 48. Photoredox-mediated synthesis of elbasvir. | |
{kind=link}
9. Desulphonation
Douglas et al. proposed a radical Smiles rearrangement in the presence of visible light to develop gem‑difluoro group found in an opioid receptor-like (ORL-1) antagonist, which is used to treat depression and obesity. The effective amalgamation of the difluoroethanol moiety with aryl and heteroaryl systems is achieved by this strategy. Thus, an array of new molecules can be synthesized. The single electron reduction of thiophene 267 produces the required difluoromethyl radical 269 recovering photocatalyst. The radical 269 undergoes Smiles rearrangement with the sulfonate group to yield 270, followed by H atom abstraction and polar SO2 extrusion to yield 268 (Scheme 49) [207].

|
Download:
|
| Scheme 49. Photocatalyzed radical Smiles rearrangement to develop gem‑difluoro group. | |
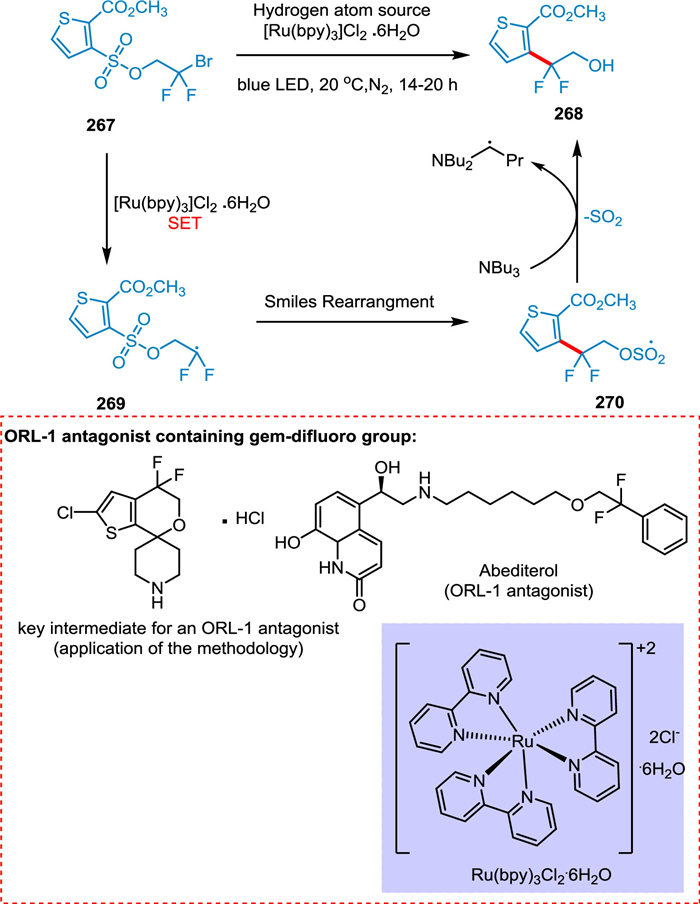{kind=link}
10. Decarboxylation
Over the past few decades, the cleavage of bonds, particularly carbon-carbon bonds, has challenged the traditional model, which has led to changes in synthetic approaches [208].
Le et al. found that, when subjected to metallaphotoredox catalysis, molecular fragments that can be easily coupled, may engage in metal insertion, decarboxylation, and recombination to make Csp2-Csp3 bonds. The acid anhydride 271 undergoes the oxidative insertion of the Ni0 complex 273 to create the equivalent acyl-carboxylate-NiII complex 274. The PC IrIII undergoes photoexcitation resulting in the corresponding *IrIII complex. The decarboxylation and SET from 274 form alkyl acyl NiIII complex 275 and IrII, respectively. The reductive elimination of embedded nickel complex results in the formation of ketone products 272. Lastly, the IrII complex and NiI species 276 would engage in a SET to simultaneously complete both catalytic cycles and restore the ground state photocatalyst IrIII and Ni0 catalyst 273 to their ground states (Scheme 50). This innovative decarboxylation-recombination approach also outlines a three-step production of the drug edivoxetine [209].

|
Download:
|
| Scheme 50. Metallaphotoredox catalysis to synthesize Csp2-Csp3 bonds. | |
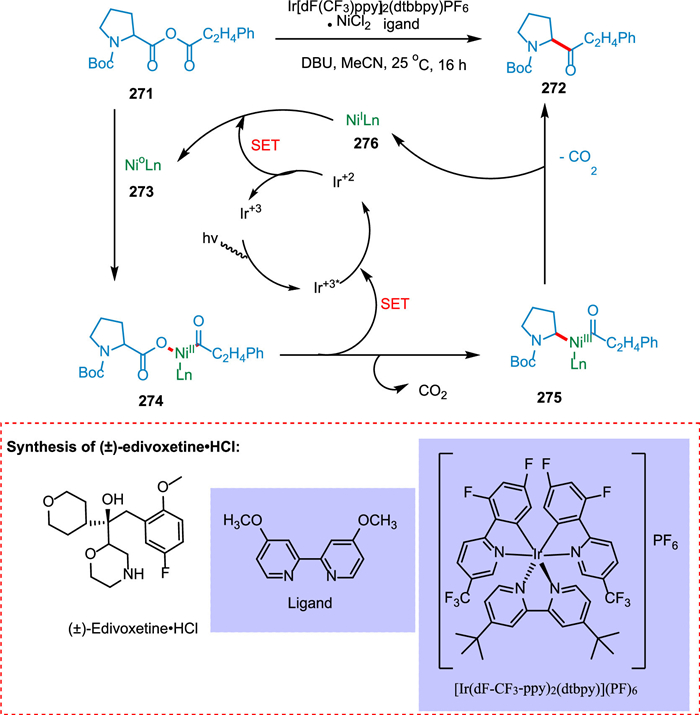{kind=link}
The cleavage of a reductive N—O bond generates nitrogen and oxygen radicals to make coupling products. This photocatalyzed approach is not much successful regardless of its major advancement due to the low atom economy. In this scenario, homolytic breaking of the N—O bond via the EnT route gives two essential radicals which is extremely helpful for circumventing the restrictions of using a single fragment. Soni et al. disclosed a novel EnT technique that couples difficult radical–radical C(sp3)-N via catalytic system reactivity modification. In the presence of photocatalyst and light, the oxime esters break homolytically, yielding oxygen radicals e.g., acyloxy radical, and nitrogen radical specie e.g., iminyl radicals, which undergo decarboxylation yielding imines. The triplet state excited photocatalyst reacts with oxime ester 277 forming excited oxime ester 281, which undergoes decarboxylation by breaking nitrogen-oxygen bond homolytically to give C 283 and N 282 radicals. These radicals couple to yield the targeted imine product 280 (Scheme 51). The solvent's nature and reaction content concentration was crucial in this cross-coupling procedure for obtaining selectivity and reactivity [210].

|
Download:
|
| Scheme 51. EnT technique couples difficult radical-radical C(sp3)-N. | |
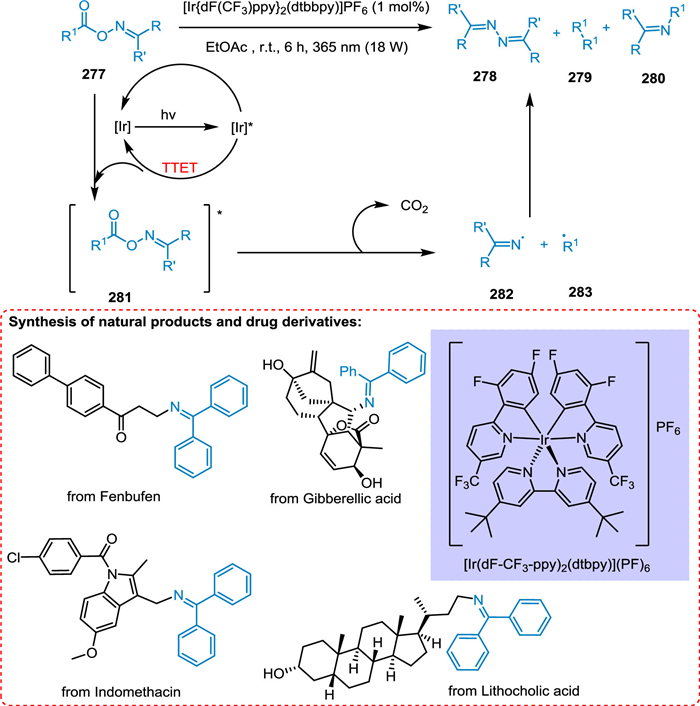{kind=link}
Hou et al. used fluorescein dimethylammonium, which is an organic dye, as a novel form of photocatalyst to carry out the decarboxylative acylation of pyridine N-oxides with α-oxocarboxylic acids. The α-oxocarboxylic acids 285 in the presence of PC goes converted into α-oxocarboxylic acids radical 288 which on decarboxylation yields benzyl radical 289. The pyridine N-oxides 284 react with benzyl radical 289 to give rise to intermediate 291, which undergoes SET resulting in cation radical intermediate 291 followed by hydrogen abstraction to yield targeted products 286 and 287 (Scheme 52). The new technique was effectively employed for synthesizing acrivastine, a vital drugintermediate [160].

|
Download:
|
| Scheme 52. Decarboxylative acylation of pyridine N-oxides with α-oxocarboxylic acids. | |
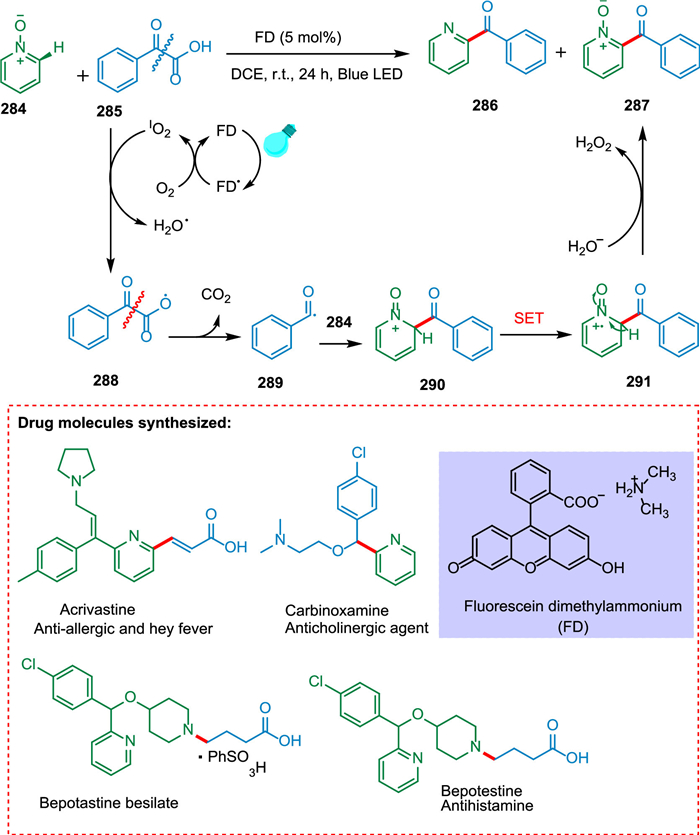{kind=link}
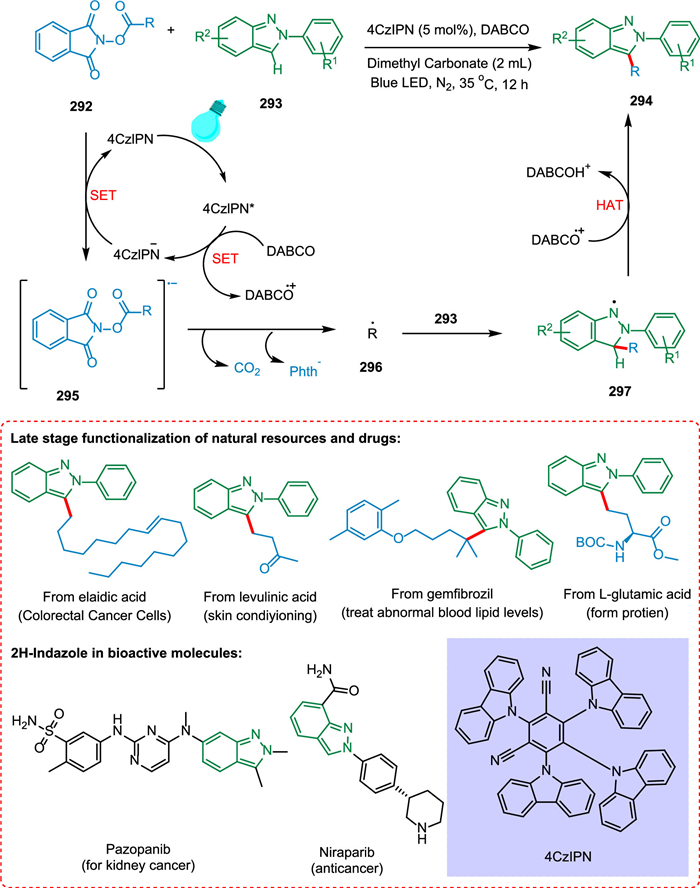
The 4CzIPN and alkyl N-hydroxyphthalimide esters 292, as the alkylating reagents, were employed by Ma et al. for alkylating 2H-indazoles without using metal. The decarboxylation and the removal of phthalimide anion convert 292 to the alkyl radical 296, which is added to the C-3 position of radical 293, forming a dearomatized radical 297. Finally, the C-3 alkylated product 294 and DABCO-H+ are produced via HAT from 297 to DABCO•+ (Scheme 53). The good functional-group compatibility makes this procedure effective to modify medicinal molecules and create new, synthetic amino acids [211].

|
Download:
|
| Scheme 53. Alkylation of 2H-indazoles without using metal. | |
{kind=link}
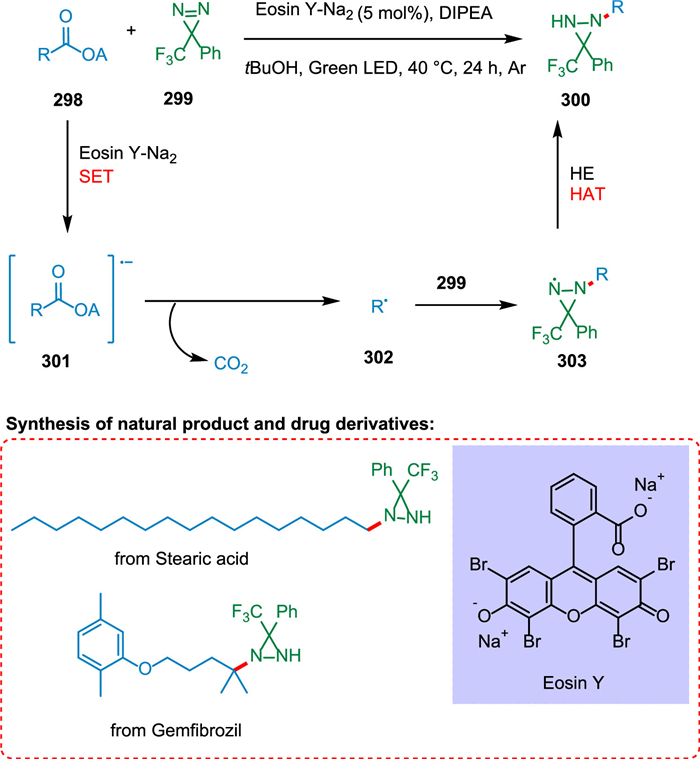
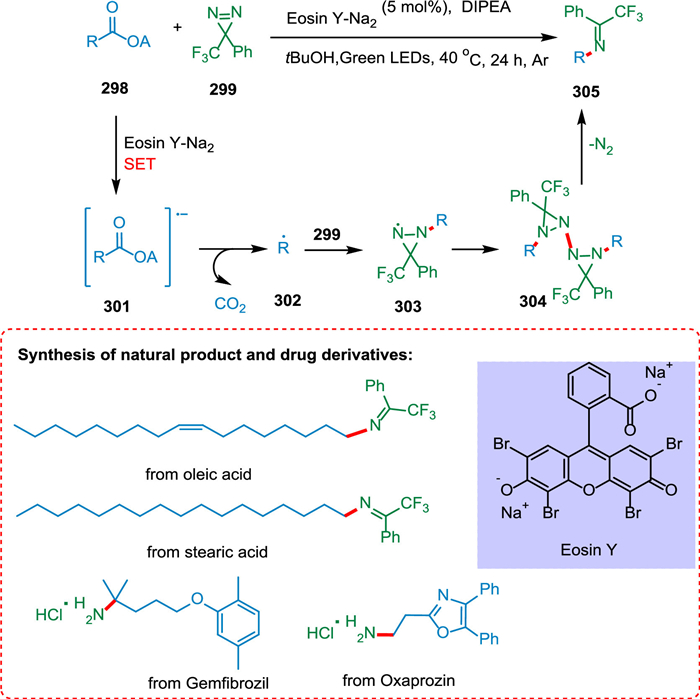
Shu et al. reported a novel divergent decarboxylative amination reaction of alkyl carboxylic acid-derived redox-active esters, allowing accessibility of amine and hydrazine from TPD [212]. The photoexcited eosin Y-Na2 (PC) is reductively quenched by DIPEA or Hantzsch ester (HE) via SET [213]. The radical anion of photocatalyst thus produces and converts ester 298 into its radical anion 301 via SET resulting in C—C bond splitting and decarboxylation forming alkyl radical 302 [214] which adds to TPD 299 to form radical species 303. These radical species take hydrogen from HE via double nitrogen transfer to yield the diaziridine products 300 (Scheme 54). Suitable hydrogen atom donors assist in hydrogen atom transfer, resulting in more diaziridine molecules. The key radical intermediate 303 can dimerize to yield the tetratetrazo intermediate 304 [215,216], which then undergoes N2 extrusion to yield two molecules of the required imines 305 (Scheme 55). This approach has broad substrate scope and has applications in drug chemistry. As a result, a wide range of imines, diaziridines, masked amines, and hydrazines are synthesized in more significant structural variations with efficacy [212].

|
Download:
|
| Scheme 54. Decarboxylative amination reaction of alkyl carboxylic acid-derived redox-active esters to synthesize diaziridines. | |
{kind=link}

|
Download:
|
| Scheme 55. Decarboxylative amination reaction of alkyl carboxylic acid-derived redox-active esters to synthesize imines. | |
{kind=link}
11. Conclusion
The field of synthetic chemistry is embracing photochemical catalytic processes, offering novel opportunities for academia and industry. This review explores the advancements in photoredox reactions having the potential for the synthesis or modification of natural products and pharmaceuticals. Dual-catalyst systems have been successfully employed to enhance photocatalysis. Despite challenges, visible light-mediated reactions continue to grow, driven by academic curiosity and the potential for discovery within the industry and successful scaling up of photoredox reactions. Collaboration between academia and industry is crucial for addressing challenges and maximizing opportunities in photocatalysis. As the field evolves, advancements in visible light catalysis offer exciting prospects for efficient and sustainable synthetic chemistry.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentThe authors express their appreciation to the Deanship of Scientific Research at King Khalid University, Saudi Arabia, for this work through Research Group Program (No. RGP-2/525/44).
| [1] |
C.K. Prier, D.A. Rankic, D.W. MacMillan, Chem. Rev. 113 (2013) 5322-5363. DOI:10.1021/cr300503r |
| [2] |
K. Kalyanasundaram, Coord. Chem. Rev. 46 (1982) 159-244. DOI:10.1016/0010-8545(82)85003-0 |
| [3] |
A. Juris, V. Balzani, F. Barigelletti, et al., Coordin. Chem. Rev. 84 (1988) 85-277. DOI:10.1016/0010-8545(88)80032-8 |
| [4] |
K.L. Wang, H.T. Ji, L.J. Ou, W.M. He, Eur. J. Org. Chem. 26 (2023) e202300752. |
| [5] |
G. Ciamician, P. Silber, Chem. Lichtwirkungen, Ber. Dtsch. Chem. Ges. 41 (1908) 1071-1080. DOI:10.1002/cber.190804101211 |
| [6] |
A. Albini, M. Fagnoni, Green Chem. 6 (2004) 1-6. DOI:10.1039/b309592d |
| [7] |
M.H. Shaw, J. Twilton, D.W. MacMillan, J. Org. Chem. 81 (2016) 6898-6926. DOI:10.1021/acs.joc.6b01449 |
| [8] |
V. Srivastava, P.K. Singh, S. Tivari, P.P. Singh, Org. Chem. Front. 9 (2022) 1485-1507. DOI:10.1039/D1QO01602D |
| [9] |
B. Dam, A.K. Sahoo, B.K. Patel, Green Chem. 24 (2022) 7122-7130. DOI:10.1039/D2GC02254K |
| [10] |
Y. Zhou, Y. Qin, Q. Wang, Z. Zhang, G. Zhu, Angew. Chem. 134 (2022) e202110864. DOI:10.1002/ange.202110864 |
| [11] |
P.P. Singh, P.K. Singh, V. Srivastava, Org. Chem. Front. 10 (2023) 216-236. DOI:10.1039/D2QO01582J |
| [12] |
M.P. Wiesenfeldt, J.A. Rossi-Ashton, I.B. Perry, et al., Nature 618 (2023) 513-518. DOI:10.1038/s41586-023-06021-8 |
| [13] |
D. Yang, Q. Yan, E. Zhu, J. Lv, W.M. He, Chin. Chem. Lett. 33 (2022) 1798-1816. DOI:10.1016/j.cclet.2021.09.068 |
| [14] |
L. Wang, P. Bao, W. Liu, et al., Chin. J. Org. Chem. 38 (2018) 3189. DOI:10.6023/cjoc201807014 |
| [15] |
L. Zheng, X. Zhuo, Y. Wang, et al., Org. Chem. Front. 9 (2022) 3012-3021. DOI:10.1039/D2QO00253A |
| [16] |
W.B. He, L.Q. Gao, X.J. Chen, Chin. Chem. Lett. 31 (2020) 1895-1898. DOI:10.1016/j.cclet.2020.02.011 |
| [17] |
Z. Guo, X. Liu, R. Bai, et al., Inorg. Chem. 60 (2021) 8672-8681. DOI:10.1021/acs.inorgchem.1c00642 |
| [18] |
K. Sun, G. Li, Y. Li, et al., Adv. Synth. Catal. 362 (2020) 1947-1954. DOI:10.1002/adsc.202000040 |
| [19] |
M.T. Pirnot, D.A. Rankic, D.B. Martin, D.W. MacMillan, Science 339 (2013) 1593-1596. DOI:10.1126/science.1232993 |
| [20] |
M. González-Esguevillas, J. Miró, J.L. Jeffrey, D.W. MacMillan, Tetrahedron 141 (2023) 133494. DOI:10.1016/j.tet.2023.133494 |
| [21] |
J.H. Shen, M. Shi, Y. Wei, Chem. Eur. J. 29 (2023) e202301157. DOI:10.1002/chem.202301157 |
| [22] |
P. Chuentragool, D. Yadagiri, T. Morita, et al., Angew. Chem. 131 (2019) 1808-1812. DOI:10.1002/ange.201812398 |
| [23] |
N. Kvasovs, V. Gevorgyan, Chem. Soc. Rev. 50 (2021) 2244-2259. DOI:10.1039/D0CS00589D |
| [24] |
M. Rivas, V. Palchykov, X. Jia, V. Gevorgyan, Nat. Rev. Chem. 6 (2022) 544-561. DOI:10.1038/s41570-022-00403-8 |
| [25] |
D.A. Nicewicz, D.W. MacMillan, Science 322 (2008) 77-80. DOI:10.1126/science.1161976 |
| [26] |
L. Shi, W. Xia, Chem. Soc. Rev. 41 (2012) 7687-7697. DOI:10.1039/c2cs35203f |
| [27] |
J.M. Narayanam, C.R. Stephenson, Chem. Soc. Rev. 40 (2011) 102-113. DOI:10.1039/B913880N |
| [28] |
T.P. Yoon, M.A. Ischay, J. Du, Nat. Chem. 2 (2010) 527-532. DOI:10.1038/nchem.687 |
| [29] |
D.M. Schultz, T.P. Yoon, Science 343 (2014) 1239176. DOI:10.1126/science.1239176 |
| [30] |
M.W. Johnson, K.I. Hannoun, Y. Tan, G.C. Fu, J.C. Peters, Chem. Sci. 7 (2016) 4091-4100. DOI:10.1039/C5SC04709A |
| [31] |
P. Marquetand, Chem. Rev. 122 (2022) 15996-15997. DOI:10.1021/acs.chemrev.2c00703 |
| [32] |
Y.M. Tian, X.N. Guo, H. Braunschweig, U. Radius, T.B. Marder, Chem. Rev. 121 (2021) 3561-3597. DOI:10.1021/acs.chemrev.0c01236 |
| [33] |
M. Fagnoni, D. Dondi, D. Ravelli, Chem. Rev. 107 (2007) 2725-2756. DOI:10.1021/cr068352x |
| [34] |
M.J. Genzink, J.B. Kidd, W.B. Swords, T.P. Yoon, Chem. Rev. 122 (2021) 1654-1716. |
| [35] |
K.L. Skubi, T.R. Blum, T.P. Yoon, Chem. Rev. 116 (2016) 10035-10074. DOI:10.1021/acs.chemrev.6b00018 |
| [36] |
T. Nevesely, M. Wienhold, J.J. Molloy, R. Gilmour, Chem. Rev. 122 (2021) 2650-2694. |
| [37] |
D.C. Blakemore, L. Castro, I. Churcher, et al., Nat. Chem. 10 (2018) 383-394. DOI:10.1038/s41557-018-0021-z |
| [38] |
K.R. Campos, P.J. Coleman, J.C. Alvarez, et al., Science 363 (2019) eaat0805. DOI:10.1126/science.aat0805 |
| [39] |
K.D.C. Lisa Candish, G.C. Cook, J.J. Douglas, et al., Chem. Rev. 122 (2022) 2907-2980. DOI:10.1021/acs.chemrev.1c00416 |
| [40] |
T. Cernak, K.D. Dykstra, S. Tyagarajan, P. Vachal, S.W. Krska, Chem. Soc. Rev. 45 (2016) 546-576. DOI:10.1039/C5CS00628G |
| [41] |
M. Moir, J.J. Danon, T.A. Reekie, M. Kassiou, Expert Opin. Drug Discov. 14 (2019) 1137-1149. DOI:10.1080/17460441.2019.1653850 |
| [42] |
L. Capaldo, L.L. Quadri, D. Ravelli, Green Chem. 22 (2020) 3376-3396. DOI:10.1039/D0GC01035A |
| [43] |
R. Cannalire, S. Pelliccia, L. Sancineto, et al., Chem. Soc. Rev. 50 (2021) 766-897. DOI:10.1039/D0CS00493F |
| [44] |
P. Bellotti, H.M. Huang, T. Faber, F. Glorius, Chem. Rev. 123 (2023) 4237-4352. DOI:10.1021/acs.chemrev.2c00478 |
| [45] |
A. Trowbridge, D. Reich, M.J. Gaunt, Nature 561 (2018) 522-527. DOI:10.1038/s41586-018-0537-9 |
| [46] |
M.D. Eastgate, M.A. Schmidt, K.R. Fandrick, Nat. Rev. Chem. 1 (2017) 0016. DOI:10.1038/s41570-017-0016 |
| [47] |
R.W. Hoffmann, Chem. Soc. Rev. 45 (2016) 577-583. DOI:10.1039/C5CS00423C |
| [48] |
A.G. Csákÿ, G. de la Herrán, M.C. Murcia, Chem. Soc. Rev. 39 (2010) 4080-4102. DOI:10.1039/b924486g |
| [49] |
J. Zhao, J.L. Brosmer, Q. Tang, et al., J. Am. Chem. Soc. 139 (2017) 9807-9810. DOI:10.1021/jacs.7b05277 |
| [50] |
M. Ikeda, K. Ohno, S.I. Mohri, M. Takahashi, Y. Tamura, J. Chem. Soc., Perkin Trans. 1 (1984) 405-412. |
| [51] |
M. Teders, C. Henkel, L. Anhäuser, et al., Nat. Chem. 10 (2018) 981-988. DOI:10.1038/s41557-018-0102-z |
| [52] |
C. Palomo, M. Oiarbide, J.M. García, Chem. Soc. Rev. 33 (2004) 65-75. DOI:10.1039/B202901D |
| [53] |
J. Xu, C. Boyer, Macromolecules 48 (2015) 520-529. DOI:10.1021/ma502460t |
| [54] |
T. Rossolini, J.A. Leitch, R. Grainger, D.J. Dixon, Org. Lett. 20 (2018) 6794-6798. DOI:10.1021/acs.orglett.8b02923 |
| [55] |
J.S. Wang, K. Wu, C. Yin, et al., Nat. Commun. 11 (2020) 4675. DOI:10.1038/s41467-020-18487-5 |
| [56] |
S. Vijayakrishnan, J.W. Ward, A.I. Cooper, ACS Catal. 12 (2022) 10057-10064. DOI:10.1021/acscatal.2c02743 |
| [57] |
F. Denes, M. Pichowicz, G. Povie, P. Renaud, Chem. Rev. 114 (2014) 2587-2693. DOI:10.1021/cr400441m |
| [58] |
G. Ciamician, Science 36 (1912) 385-394. DOI:10.1126/science.36.926.385 |
| [59] |
J. Metternich, R. Gilmour, Synlett 27 (2016) 2541-2552. DOI:10.1055/s-0036-1588621 |
| [60] |
Z. Lu, T.P. Yoon, Angew. Chem. 124 (2012) 10475-10478. DOI:10.1002/ange.201204835 |
| [61] |
T.R. Blum, Z.D. Miller, D.M. Bates, I.A. Guzei, T.P. Yoon, Science 354 (2016) 1391-1395. DOI:10.1126/science.aai8228 |
| [62] |
E.R. Welin, C. Le, D.M. Arias-Rotondo, J.K. McCusker, D.W. MacMillan, Science 355 (2017) 380-385. DOI:10.1126/science.aal2490 |
| [63] |
J. Mayol-Llinas, A. Nelson, W. Farnaby, A. Ayscough, Drug Discov. Today 22 (2017) 965-969. DOI:10.1016/j.drudis.2017.01.008 |
| [64] |
V. Lahmy, R. Long, D. Morin, V. Villard, T. Maurice, Front. Cell. Neurosci. 8 (2015) 463. |
| [65] |
D.P. Marciano, M.R. Chang, C.A. Corzo, et al., Cell Metab. 19 (2014) 193-208. DOI:10.1016/j.cmet.2013.12.009 |
| [66] |
P. Ruiz-Castillo, S.L. Buchwald, Chem. Rev. 116 (2016) 12564-12649. DOI:10.1021/acs.chemrev.6b00512 |
| [67] |
S.B. Herzon, J.F. Hartwig, J. Am. Chem. Soc. 130 (2008) 14940-14941. DOI:10.1021/ja806367e |
| [68] |
L. Huang, M. Arndt, K.t. Gooßen, H. Heydt, L.J. Goossen, Chem. Rev. 115 (2015) 2596-2697. DOI:10.1021/cr300389u |
| [69] |
P. Kalck, M. Urrutigoity, Chem. Rev. 118 (2018) 3833-3861. DOI:10.1021/acs.chemrev.7b00667 |
| [70] |
F. Perez, S. Oda, L.M. Geary, M.J. Krische, Top. Curr. Chem. 374 (2016) 365-387. |
| [71] |
M. Chilamari, J.R. Immel, S. Bloom, ACS Catal. 10 (2020) 12727-12737. DOI:10.1021/acscatal.0c03422 |
| [72] |
G. Wuitschik, E.M. Carreira, B.r. Wagner, et al., J. Med. Chem. 53 (2010) 3227-3246. DOI:10.1021/jm9018788 |
| [73] |
D. Ravelli, M. Zoccolillo, M. Mella, M. Fagnoni, Adv. Synth. Catal. 356 (2014) 2781-2786. DOI:10.1002/adsc.201400027 |
| [74] |
S.H. Oh, Y.R. Malpani, N. Ha, Y.S. Jung, S.B. Han, Org. Lett. 16 (2014) 1310-1313. DOI:10.1021/ol403716t |
| [75] |
J. Yang, Z. Sun, K. Yan, et al., Green Chem. 23 (2021) 2756-2762. DOI:10.1039/D1GC00379H |
| [76] |
Y.H. Lu, C. Wu, J.C. Hou, et al., ACS Catal. 13 (2023) 13071-13076. DOI:10.1021/acscatal.3c02268 |
| [77] |
H.T. Ji, K.L. Wang, W.T. Ouyang, et al., Green Chem. 25 (2023) 7983-7987. DOI:10.1039/D3GC02575F |
| [78] |
P.V. Fish, A.D. Brown, E. Evrard, L.R. Roberts, Bioorg. Med. Chem. Lett. 19 (2009) 1871-1875. DOI:10.1016/j.bmcl.2009.02.071 |
| [79] |
A. Brown, T.B. Brown, A. Calabrese, et al., Bioorg. Med. Chem. Lett. 20 (2010) 516-520. DOI:10.1016/j.bmcl.2009.11.097 |
| [80] |
M. Maetani, J. Zoller, B. Melillo, et al., J. Am. Chem. Soc. 139 (2017) 11300-11306. DOI:10.1021/jacs.7b06994 |
| [81] |
M.R. Becker, E.R. Wearing, C.S. Schindler, Nat. Chem. 12 (2020) 898-905. DOI:10.1038/s41557-020-0541-1 |
| [82] |
F. Wang, C. Chen, G. Deng, C. Xi, J. Org. Chem. 77 (2012) 4148-4151. DOI:10.1021/jo300250x |
| [83] |
F. Ke, Y. Qu, Z. Jiang, et al., Org. Lett. 13 (2011) 454-457. DOI:10.1021/ol102784c |
| [84] |
Z. Chen, Q. Yan, Z. Liu, Y. Zhang, Chem. Eur. J. 20 (2014) 17635-17639. DOI:10.1002/chem.201405057 |
| [85] |
M.E. Daub, M. Ehrenshaft, Annu. Rev. Phytopathol. 38 (2000) 461-490. DOI:10.1146/annurev.phyto.38.1.461 |
| [86] |
Y. Zhang, Y. Cao, L. Lu, et al., J. Org. Chem. 84 (2019) 7711-7721. DOI:10.1021/acs.joc.9b00545 |
| [87] |
J.B. Metternich, R. Gilmour, J. Org. Chem. 138 (2016) 1040-1045. |
| [88] |
G.K. Jana, S. Paul, S. Sinha, Org. Prep. Proc. Int. 43 (2011) 541-573. DOI:10.1080/00304948.2011.629563 |
| [89] |
X.Y. Liu, Y. Qin, Acc. Chem. Res. 52 (2019) 1877-1891. DOI:10.1021/acs.accounts.9b00246 |
| [90] |
Z. Zhang, D. Yi, M. Zhang, et al., ACS Catal. 10 (2020) 10149-10156. DOI:10.1021/acscatal.0c01841 |
| [91] |
M.S. Oderinde, J. Kempson, D. Smith, et al., Eur. J. Org. Chem. 2020 (2020) 41-46. DOI:10.1002/ejoc.201901482 |
| [92] |
Y. Bansal, O.J.B. Silakari, Bioorg. Med. Chem. 20 (2012) 6208-6236. DOI:10.1016/j.bmc.2012.09.013 |
| [93] |
P.A. Renhowe, S. Pecchi, C.M. Shafer, et al., J. Med. Chem. 52 (2009) 278-292. DOI:10.1021/jm800790t |
| [94] |
C. Zhang, B. Zhong, S. Yang, et al., Bioorg. Med. Chem. 23 (2015) 3774-3780. DOI:10.1016/j.bmc.2015.03.085 |
| [95] |
P.D. Verdouw, J.M. Hartog, D.J. Duncker, W. Roth, P.R. Saxena, Eur. J. Pharmacol. 126 (1986) 21-30. DOI:10.1016/0014-2999(86)90733-8 |
| [96] |
R. Mocharla, H. Mocharla, M.E. Hodes, Nucleic Acids Res. 15 (1987) 10589. DOI:10.1093/nar/15.24.10589 |
| [97] |
Y. Kim, M.R. Kumar, N. Park, Y. Heo, S. Lee, J. Org. Chem. 76 (2011) 9577-9583. DOI:10.1021/jo2019416 |
| [98] |
K. Bahrami, M.M. Khodaei, F. Naali, J. Org. Chem. 73 (2008) 6835-6837. DOI:10.1021/jo8010232 |
| [99] |
J. Kovvuri, B. Nagaraju, A. Kamal, A.K. Srivastava, ACS Comb. Sci. 18 (2016) 644-650. DOI:10.1021/acscombsci.6b00107 |
| [100] |
J.W. Tucker, J.M. Narayanam, S.W. Krabbe, C.R. Stephenson, Org. Lett. 12 (2010) 368-371. DOI:10.1021/ol902703k |
| [101] |
H. Huang, M. Yu, X. Su, et al., J. Org. Chem. 83 (2018) 2425-2437. DOI:10.1021/acs.joc.7b03017 |
| [102] |
P.D. Bass, D.A. Gubler, T.C. Judd, R.M. Williams, Chem. Rev. 113 (2013) 6816-6863. DOI:10.1021/cr3001059 |
| [103] |
T. Sasase, H. Morinaga, T. Abe, et al., Diabet. Obes. Metab. 11 (2009) 1084-1087. DOI:10.1111/j.1463-1326.2009.01082.x |
| [104] |
V.M. Dembitsky, J. Nat. Med. 62 (2008) 1-33. |
| [105] |
S. Ren, F. Pan, W. Zhang, G.W. Rao, Curr. Med. Chem. 29 (2022) 4113-4135. DOI:10.2174/0929867329666220105120722 |
| [106] |
E. Lee-Ruff, G. Mladenova, Chem. Rev. 103 (2003) 1449-1484. DOI:10.1021/cr010013a |
| [107] |
Y. Xu, M.L. Conner, M.K. Brown, Angew. Chem. Int. Ed. 54 (2015) 11918-11928. DOI:10.1002/anie.201502815 |
| [108] |
T. Sravanthi, S. Manju, Eur. J. Pharm. Sci. 91 (2016) 1-10. DOI:10.1016/j.ejps.2016.05.025 |
| [109] |
N. Chadha, O. Silakari, Eur. J. Med. Chem. 134 (2017) 159-184. DOI:10.1016/j.ejmech.2017.04.003 |
| [110] |
M.S. Oderinde, A. Ramirez, T.M. Dhar, et al., J. Org. Chem. 86 (2020) 1730-1747. |
| [111] |
L.R. Orgren, E.E. Maverick, C.C. Marvin, J. Org. Chem. 80 (2015) 12635-12640. DOI:10.1021/acs.joc.5b02199 |
| [112] |
A. Brossi, L. Chopard-dit-Jean, O. Schnider, Helv. Chim. Acta 41 (1958) 1793-1806. DOI:10.1002/hlca.19580410634 |
| [113] |
B. Giese, B. Kopping, T. Göbel, et al., Org. React. 48 (2004) 301-856. |
| [114] |
Y. Liu, M. Zhang, C.H. Tung, Y. Wang, ACS Catal. 6 (2016) 8389-8394. DOI:10.1021/acscatal.6b03076 |
| [115] |
Z. Li, H. Song, R. Guo, et al., Green Chem. 21 (2019) 3602-3605. DOI:10.1039/C9GC01359H |
| [116] |
X. Zhuang, X. Shi, R. Zhu, et al., Org. Chem. Front. 8 (2021) 736-742. DOI:10.1039/D0QO01188F |
| [117] |
S. Özden, D. Atabey, S. Yıldız, H. Göker, Bioorg. Med. Chem. 13 (2005) 1587-1597. DOI:10.1016/j.bmc.2004.12.025 |
| [118] |
J.Y. Chai, B.K. Jung, S.J. Hong, Korean J. Parasitol. 59 (2021) 189. DOI:10.3347/kjp.2021.59.3.189 |
| [119] |
S.M. Sondhi, S. Rajvanshi, M. Johar, et al., Eur. J. Med. Chem. 37 (2002) 835-843. DOI:10.1016/S0223-5234(02)01403-4 |
| [120] |
X.Z. Guo, L. Shi, R. Wang, et al., Bioorg. Med. Chem. 16 (2008) 10301-10310. DOI:10.1016/j.bmc.2008.10.040 |
| [121] |
A. Chimirri, A. De Sarro, G. De Sarro, R. Gitto, M. Zappala, Farmaco 56 (2001) 821-826. DOI:10.1016/S0014-827X(01)01147-8 |
| [122] |
A. Lee-Dutra, K.L. Arienti, D.J. Buzard, et al., Bioorg. Med. Chem. Lett. 16 (2006) 6043-6048. DOI:10.1016/j.bmcl.2006.08.117 |
| [123] |
M.J. O'Neil, M. Smith, P. Heckelman, The Merck Index, 13th Edition, Merck and Co, Inc NJ, 2001, p. 1785.
|
| [124] |
I. Siddiqui, F. Ibad, A. Ibad, M.A. Waseem, G. Watal, Tetrahedron Lett. 57 (2016) 5-10. DOI:10.1016/j.tetlet.2015.10.042 |
| [125] |
A. Armstrong, J.C. Collins, Angew. Chem. Int. Ed. 49 (2010) 2282-2285. DOI:10.1002/anie.200906750 |
| [126] |
A. Gauld, STEPS: suvorexant (Belsomra) for insomnia, Am. Fam. Phys. 93 (2016) 1016. |
| [127] |
V.S. Patil, D.V. Patil, S.S. Potdar, Org. Commun. 15 (2022) 71-80. |
| [128] |
V.K. Yadav, V.P. Srivastava, L.D.S. Yadav, Tetrahedron Lett. 57 (2016) 155-158. DOI:10.1016/j.tetlet.2015.11.089 |
| [129] |
S. Zykova, S. Shurov, V. Talismanov, et al., J. Pharm. Sci. Res. 10 (2018) 947-949. |
| [130] |
Z.P. Ye, Y.Z. Hu, J.P. Guan, et al., Org. Lett. 23 (2021) 4754-4758. DOI:10.1021/acs.orglett.1c01477 |
| [131] |
G. Zhou, X. Deng, C. Pan, et al., Chem. Comm. 56 (2020) 12546-12549. DOI:10.1039/D0CC04471G |
| [132] |
C. Russo, R. Cannalire, P. Luciano, et al., Synthesis 53 (2021) 4419-4427. DOI:10.1055/a-1543-3924 |
| [133] |
G.E. Crisenza, P. Melchiorre, Nat. Commun. 11 (2020) 1-4. DOI:10.1038/s41467-019-13993-7 |
| [134] |
V.R. Solomon, H. Lee, Curr. Med. Chem. 18 (2011) 1488-1508. DOI:10.2174/092986711795328382 |
| [135] |
J.X. Xu, N.L. Pan, J.X. Chen, J.W. Zhao, J. Org. Chem. 86 (2021) 10747-10754. DOI:10.1021/acs.joc.1c01386 |
| [136] |
J. Ma, F. Schäfers, C. Daniliuc, et al., Angew. Chem. 132 (2020) 9726-9732. DOI:10.1002/ange.202001200 |
| [137] |
C.E. Knappke, S. Grupe, D. Gärtner, et al., Chem. Eur. J. 20 (2014) 6828-6842. DOI:10.1002/chem.201402302 |
| [138] |
D.A. Everson, D.J. Weix, J. Org. Chem. 79 (2014) 4793-4798. DOI:10.1021/jo500507s |
| [139] |
P.J. Xia, Z.P. Ye, D. Song, et al., Chem. Comm. 55 (2019) 2712-2715. DOI:10.1039/C8CC09385G |
| [140] |
X. Tian, Y. Guo, W. An, et al., Nat. Commun. 13 (2022) 6186. DOI:10.1038/s41467-022-33778-9 |
| [141] |
S. Tripathi, L.D.S. Yadav, New J. Chem. 42 (2018) 3765-3769. DOI:10.1039/C7NJ04578F |
| [142] |
R.W. Gantt, P. Peltier-Pain, J.S. Thorson, Nat. Prod. Rep. 28 (2011) 1811-1853. DOI:10.1039/c1np00045d |
| [143] |
Y. Yang, B. Yu, Chem. Rev. 117 (2017) 12281-12356. DOI:10.1021/acs.chemrev.7b00234 |
| [144] |
Q. Wu, J.G. Cho, D.S. Lee, et al., Carbohydr. Res. 372 (2013) 9-14. DOI:10.1016/j.carres.2012.09.015 |
| [145] |
W. Disadee, C. Mahidol, P. Sahakitpichan, et al., Phytochemistry 74 (2012) 115-122. DOI:10.1016/j.phytochem.2011.11.001 |
| [146] |
T. Kawamata, M. Nagatomo, M. Inoue, J. Am. Chem. Soc. 139 (2017) 1814-1817. DOI:10.1021/jacs.6b13263 |
| [147] |
S.O. Badir, A. Dumoulin, J.K. Matsui, G.A. Molander, Angew. Chem. Int. Ed. 57 (2018) 6610-6613. DOI:10.1002/anie.201800701 |
| [148] |
X. Wang, J. Dong, Y. Liu, H. Song, Q. Wang, Chin. Chem. Lett. 32 (2021) 3027-3030. DOI:10.1016/j.cclet.2021.03.070 |
| [149] |
M.P. DeMartino, K. Chen, P.S. Baran, J. Am. Chem. Soc. 130 (2008) 11546-11560. DOI:10.1021/ja804159y |
| [150] |
Y. Kuang, K. Wang, X. Shi, et al., Angew. Chem. 131 (2019) 17015-17019. DOI:10.1002/ange.201910414 |
| [151] |
S. Li, P.W. Davies, W. Shu, Chem. Sci. 13 (2022) 6636-6641. DOI:10.1039/D2SC01905A |
| [152] |
N.A. Romero, K.A. Margrey, N.E. Tay, D.A. Nicewicz, Science 349 (2015) 1326-1330. DOI:10.1126/science.aac9895 |
| [153] |
J.K. Matsui, D.N. Primer, G.A. Molander, Chem. Sci. 8 (2017) 3512-3522. DOI:10.1039/C7SC00283A |
| [154] |
H. Liu, L. Ge, D.X. Wang, N. Chen, C. Feng, Angew. Chem. 131 (2019) 3958-3962. DOI:10.1002/ange.201814308 |
| [155] |
M.A. Miranda, M.L. Marin, Curr. Opin. Green Sustain. Chem. 6 (2017) 139-149. DOI:10.1016/j.cogsc.2017.05.001 |
| [156] |
L. Guillemard, N. Kaplaneris, L. Ackermann, M.J. Johansson, Nat. Rev. Chem. 5 (2021) 522-545. DOI:10.1038/s41570-021-00300-6 |
| [157] |
B. Liu, L. Yang, P. Li, F. Wang, X. Li, Org. Chem. Front. 8 (2021) 1085-1101. DOI:10.1039/D0QO01159B |
| [158] |
K.J. Jiao, Y.K. Xing, Q.L. Yang, H. Qiu, T.S. Mei, Acc. Chem. Res. 53 (2020) 300-310. DOI:10.1021/acs.accounts.9b00603 |
| [159] |
S. Vidyacharan, B.T. Ramanjaneyulu, S. Jang, D.P. Kim, ChemSusChem 12 (2019) 2581-2586. DOI:10.1002/cssc.201900736 |
| [160] |
C. Hou, S. Sun, Z. Liu, et al., Adv. Synth. Catal. 363 (2021) 2806-2812. DOI:10.1002/adsc.202100168 |
| [161] |
H.Y. Song, F. Xiao, J. Jiang, et al., Chin. Chem. Lett. (2023) 108509. |
| [162] |
H. Xu, X. Li, J. Ma, et al., Chin. Chem. Lett. (2023) 108403. |
| [163] |
H. Cerecetto, A. Gerpe, M. Gonzalez, V.J. Aran, C.O. de Ocariz, Mini-Rev. Med. Chem. 5 (2005) 869-878. DOI:10.2174/138955705774329564 |
| [164] |
X. Li, S. Chu, V.A. Feher, et al., J. Med. Chem. 46 (2003) 5663-5673. DOI:10.1021/jm0302039 |
| [165] |
G. Picciola, F. Ravenna, G. Carenini, P. Gentili, M. Riva, Il Farmaco, Edizione Sci. 36 (1981) 1037-1056. |
| [166] |
W. Han, J.C. Pelletier, C.N. Hodge, Bioorg. Med. Chem. Lett. 8 (1998) 3615-3620. DOI:10.1016/S0960-894X(98)00659-3 |
| [167] |
J. Liu, C. Qian, Y. Zhu, et al., Bioorg. Med. Chem. 26 (2018) 747-757. DOI:10.1016/j.bmc.2017.12.041 |
| [168] |
S.V. Keisner, S.R. Shah, Drugs 71 (2011) 443-454. |
| [169] |
A. Unsinn, P. Knochel, Chem. Comm. 48 (2012) 2680-2682. DOI:10.1039/c2cc17804d |
| [170] |
S.A. Ohnmacht, A.J. Culshaw, M.F. Greaney, Org. Lett. 12 (2010) 224-226. DOI:10.1021/ol902537d |
| [171] |
X. Ding, J. Bai, H. Wang, et al., Tetrahedron 73 (2017) 172-178. DOI:10.1016/j.tet.2016.11.066 |
| [172] |
G. Bogonda, H.Y. Kim, K. Oh, Org. Lett. 20 (2018) 2711-2715. DOI:10.1021/acs.orglett.8b00920 |
| [173] |
W. Zhang, X.X. Xiang, J. Chen, et al., Nat. Commun. 11 (2020) 1-10. DOI:10.1038/s41467-019-13993-7 |
| [174] |
P. Nuhant, M.S. Oderinde, J. Genovino, et al., Angew. Chem. Int. Ed. 56 (2017) 15309-15313. DOI:10.1002/anie.201707958 |
| [175] |
Q.Q. Han, D.M. Chen, Z.L. Wang, et al., Chin. Chem. Lett. 32 (2021) 2559-2561. DOI:10.1016/j.cclet.2021.02.018 |
| [176] |
M.D. Tzirakis, I.N. Lykakis, M. Orfanopoulos, Chem. Soc. Rev. 38 (2009) 2609-2621. DOI:10.1039/b812100c |
| [177] |
D. Mazzarella, A. Pulcinella, L. Bovy, R. Broersma, T. Noël, Angew. Chem. Int. Ed. 60 (2021) 21277-21282. DOI:10.1002/anie.202108987 |
| [178] |
Y.W. Zheng, R. Narobe, K. Donabauer, S. Yakubov, B. König, ACS Catal. 10 (2020) 8582-8589. DOI:10.1021/acscatal.0c01924 |
| [179] |
J.Y. Dong, H.P. He, Y.M. Shen, K.Q. Zhang, J. Nat. Prod. 68 (2005) 1510-1513. DOI:10.1021/np0502241 |
| [180] |
C. Takahashi, A. Numata, E. Matsumura, et al., J. Antibiot. 47 (1994) 1242-1249. DOI:10.7164/antibiotics.47.1242 |
| [181] |
C.J. Zheng, C.J. Kim, K.S. Bae, Y.H. Kim, W.G. Kim, J. Nat. Prod. 69 (2006) 1816-1819. DOI:10.1021/np060348t |
| [182] |
L. Furst, J.M. Narayanam, C.R. Stephenson, Angew. Chem. Int. Ed. 50 (2011) 9655. DOI:10.1002/anie.201103145 |
| [183] |
D.A. Nagib, D.W. MacMillan, Nature 480 (2011) 224-228. DOI:10.1038/nature10647 |
| [184] |
I.B. Seiple, S. Su, R.A. Rodriguez, et al., J. Am. Chem. Soc. 132 (2010) 13194-13196. DOI:10.1021/ja1066459 |
| [185] |
R. Pschorr, Ber. Dtsch. Chem. Ges. 29 (1896) 496-501. DOI:10.1002/cber.18960290198 |
| [186] |
D. Xue, Z.H. Jia, C.J. Zhao, et al., Chem. Eur. J. 20 (2014) 2960-2965. DOI:10.1002/chem.201304120 |
| [187] |
E.R. Welin, A.A. Warkentin, J.C. Conrad, D.W. MacMillan, Angew. Chem. Int. Ed. 54 (2015) 9668-9672. DOI:10.1002/anie.201503789 |
| [188] |
A. Singh, A. Arora, J.D. Weaver, Org. Lett. 15 (2013) 5390-5393. DOI:10.1021/ol402751j |
| [189] |
D.A. DiRocco, K. Dykstra, S. Krska, et al., Angew. Chem. Int. Ed. 53 (2014) 4802-4806. DOI:10.1002/anie.201402023 |
| [190] |
G. Lei, M. Xu, R. Chang, I. Funes-Ardoiz, J. Ye, J. Am. Chem. Soc. 143 (2021) 11251-11261. DOI:10.1021/jacs.1c05852 |
| [191] |
J.M. Mayer, Acc. Chem. Res. 44 (2011) 36-46. DOI:10.1021/ar100093z |
| [192] |
M. Costas, M. Bietti, Acc. Chem. Res. 51 (2018) 2601-2602. DOI:10.1021/acs.accounts.8b00525 |
| [193] |
C.R. Stephenson, T.P. Yoon, D.W. MacMillan, Visible Light Photocatalysis In Organic Chemistry, John Wiley & Sons, 2018.
|
| [194] |
D. Staveness, I. Bosque, C.R. Stephenson, Acc. Chem. Res. 49 (2016) 2295-2306. DOI:10.1021/acs.accounts.6b00270 |
| [195] |
B.P. Roberts, Chem. Soc. Rev. 28 (1999) 25-35. DOI:10.1039/a804291h |
| [196] |
S. Cao, W. Hong, Z. Ye, L. Gong, Nat. Commun. 12 (2021) 2377. DOI:10.1038/s41467-021-22690-3 |
| [197] |
H. Du, C. Fairbridge, H. Yang, Z. Ring, Appl. Catal. A: Gen. 294 (2005) 1-21. DOI:10.1016/j.apcata.2005.06.033 |
| [198] |
W. Zhang, Y.L. Pan, C. Yang, X. Li, B. Wang, Org. Chem. Front. 6 (2019) 2765-2770. DOI:10.1039/C9QO00625G |
| [199] |
X. Zhu, X. Song, X. Li, et al., Adv. Synth. Catal. 364 (2022) 4384-4391. DOI:10.1002/adsc.202201143 |
| [200] |
W. Ou, G. Zhang, J. Wu, C. Su, ACS Catal. 9 (2019) 5178-5183. DOI:10.1021/acscatal.9b00693 |
| [201] |
X.Z. Fan, J.W. Rong, H.L. Wu, et al., Angew. Chem. 130 (2018) 8650-8654. DOI:10.1002/ange.201803220 |
| [202] |
I. Funes-Ardoiz, F. Maseras, ACS Catal. 8 (2018) 1161-1172. DOI:10.1021/acscatal.7b02974 |
| [203] |
H.Y. Song, J. Jiang, C. Wu, et al., Green Chem. 25 (2023) 3292-3296. DOI:10.1039/D2GC04843D |
| [204] |
J. Li, J. Chen, R. Sang, et al., Nat. Chem. 12 (2020) 56-62. DOI:10.1038/s41557-019-0353-3 |
| [205] |
X. Sun, J. Chen, T. Ritter, Nat. Chem. 10 (2018) 1229-1233. DOI:10.1038/s41557-018-0142-4 |
| [206] |
H.G. Yayla, F. Peng, I.K. Mangion, et al., Chem. Sci. 7 (2016) 2066-2073. DOI:10.1039/C5SC03350K |
| [207] |
J.J. Douglas, H. Albright, M.J. Sevrin, K.P. Cole, C.R. Stephenson, Angew. Chem. Int. Ed. 54 (2015) 14898-14902. DOI:10.1002/anie.201507369 |
| [208] |
Y.F. Liang, M. Bilal, L.Y. Tang, et al., Chem. Rev. 123 (2023) 12313-12370. DOI:10.1021/acs.chemrev.3c00219 |
| [209] |
C.C. Le, D.W. MacMillan, J. Am. Chem. Soc. 137 (2015) 11938-11941. DOI:10.1021/jacs.5b08304 |
| [210] |
V.K. Soni, S. Lee, J. Kang, et al., ACS Catal. 9 (2019) 10454-10463. DOI:10.1021/acscatal.9b03435 |
| [211] |
C. Ma, Z. Feng, J. Li, et al., Org. Chem. Front. 8 (2021) 3286-3291. DOI:10.1039/D1QO00064K |
| [212] |
X. Shu, R. Xu, S. Liao, Sci. China Chem. 64 (2021) 1756-1762. DOI:10.1007/s11426-021-1048-4 |
| [213] |
R. Xu, T. Xu, M. Yang, T. Cao, S. Liao, Nat. Commun. 10 (2019) 3752. DOI:10.1038/s41467-019-11805-6 |
| [214] |
K. Okada, K. Okamoto, M. Oda, J. Am. Chem. Soc. 110 (1988) 8736-8738. DOI:10.1021/ja00234a047 |
| [215] |
D.M. Gale, W.J. Middleton, C.G. Krespan, J. Am. Chem. Soc. 88 (1966) 3617-3623. DOI:10.1021/ja00967a026 |
| [216] |
D.H. Barton, J.C. Jaszberenyi, E.A. Theodorakis, J. Reibenspies, J. Am. Chem. Soc. 115 (1993) 8050-8059. DOI:10.1021/ja00071a017 |