 2024, Vol. 35
2024, Vol. 35 


b Soochow Institute of Energy and Material Innovations, College of Energy, Jiangsu Key Laboratory of Advanced Carbon Materials and Wearable Energy Technologies, Soochow University, Soochow 215006, China;
c Jiangsu Key Laboratory of Advanced Negative Carbon Technologies, Soochow University, Soochow 215123, China;
d School of Chemistry and Chemical Engineering, Shihezi University, Shihezi 832003, China
The increase in atmospheric carbon dioxide levels has been identified as a significant contributing factor to the deterioration of the global climate and the acceleration of global warming. According to reports, atmospheric CO2 concentrations have increased from 280 ppm to 415 ppm over the previous two centuries, and predictions suggest they could reach over 600 ppm by 2100. Such a trend is expected to disrupt the carbon equilibrium between the atmosphere and ecosystems [1,2]. Further, under a no-policy baseline scenario [3], scientific projections indicate that the continued emission of CO2 will lead to a global temperature increase of over 4 ℃ by 2100. In light of these consequences, there is an urgent need to explore CO2 utilization and reduction strategies.
The electrocatalytic reduction of CO2 (CO2RR) has the potential to mitigate climate change by transforming carbon dioxide into valuable chemicals, thereby reducing emissions and closing the anthropogenic carbon cycle [4,5]. Many prior investigations have focused on designing active sites for electrocatalysts, including the regulation of crystal planes [6–8], defect engineering [9–14], oxidation state [15–18], surface modification [19–21], and metal single-atoms [22,23]. Additionally, researchers have discovered that the electrolyte environment plays a crucial role in achieving highly efficient and selective CO2RR. Since the electrocatalytic reaction occurs at the electrode-electrolyte interface, electrolytes are thought to be substantially engaged in the CO2RR process via interactions with catalyst surfaces, intermediates, and reactants. An electric double layer (EDL) can be observed at the juncture where the electrode and the electrolyte intersect. The outer Helmholtz plane (OHP) is populated by hydrated ions, while the inner Helmholtz plane (IHP) is predominantly inhabited by covalently bonded species and reaction intermediates [24]. During electrolysis, the working electrode has a potential that is more negative than the point of zero charges, causing hydrated cations to be drawn to the electrode by electrostatic forces and increasing the population of cations on OHP. The aggregation of cations within the OHP notably modulates the microenvironment existing at the electrode/electrolyte interface.
The cation effect on CO2RR was initially discovered in the late 1960s when Paik and colleagues observed that the overpotential for CO2RR to HCOOH decreased and the current density increased with a switch to a Na+ or tetraethylammonium-based electrolyte [25]. Since then, a growing body of research has demonstrated that the cation species in the electrolyte environment profoundly influence the efficiency of CO2RR. For example, Huang et al. [26] achieved a significant breakthrough by introducing K+ into an acidic electrolyte, which facilitated the efficient reduction of CO2 to formic acid (HCOOH). This process yielded a current density of −237.1 mA/cm2, and exhibited an impressively beyond 90% Faradaic efficiency (FE). Ager et al. have discerned a remarkable escalation, exceeding an order of magnitude, in the generation rate of multi-carbon (C2+) products during CO2RR upon transitioning from K+-containing electrolyte to one containing Cs+. This observation implies a noteworthy role of Cs+ in augmenting the efficiency of the CO2RR process [27]. Kyriacou et al. [28] observed that the CO2 reduction rate increased with increasing cation surface charge, with the CO2 reduction rate of the salt system including La3+ being twice that of the salt system containing Na+ at low potentials. Sargent et al. [29] employed a cation-enhancing strategy to improve the CO2 activation process in a strongly acidic environment. Their findings revealed that the incorporation of cations (K+) led to a substantial suppression of the Hydrogen Evolution Reaction (HER) process. This, in turn, resulted in an increase in the yield of C2+ products, which was directly proportional to the concentration of the introduced cation (K+). Though great advances have been made in understanding the cation effect, it remains highly controversial how cations affect the local electrolyte environment in CO2RR. This is attributed to the complexity of the electrode/electrolyte interfacial environment determined by the cationic effect, as specific cationic constituents within the electrolyte playing a pivotal role in reshaping the interfacial topology and modulating the adsorptive characteristics of the intermediates. To date, the existing models of cation effects have only been able to rationalize various experimentally observed trends and lack general predictive capabilities. In light of this, we present an exhaustive overview of prevailing comprehension concerning cationic effects in CO2RR, meticulously analyzing the topic from both theoretical and experimental perspectives. Our discussion covers a range of factors, including interfacial electric field, local pH, stabilization of key intermediates, and interfacial water environment. Our focus is on elucidating the fundamental principles governing the interaction of cations with CO2RR catalytic process and the resulting impact on the reaction kinetics and selectivity. Moreover, we outline valuable guidance for harnessing cationic species in the design of high-performance CO2RR systems, coupled with an appraisal of the existing opportunities and challenges in this domain. Through this effort, we hope to advance the understanding of the intricate interplay between cationic species and CO2RR mechanisms, thereby facilitating the development of universally predictive models for cation effects.
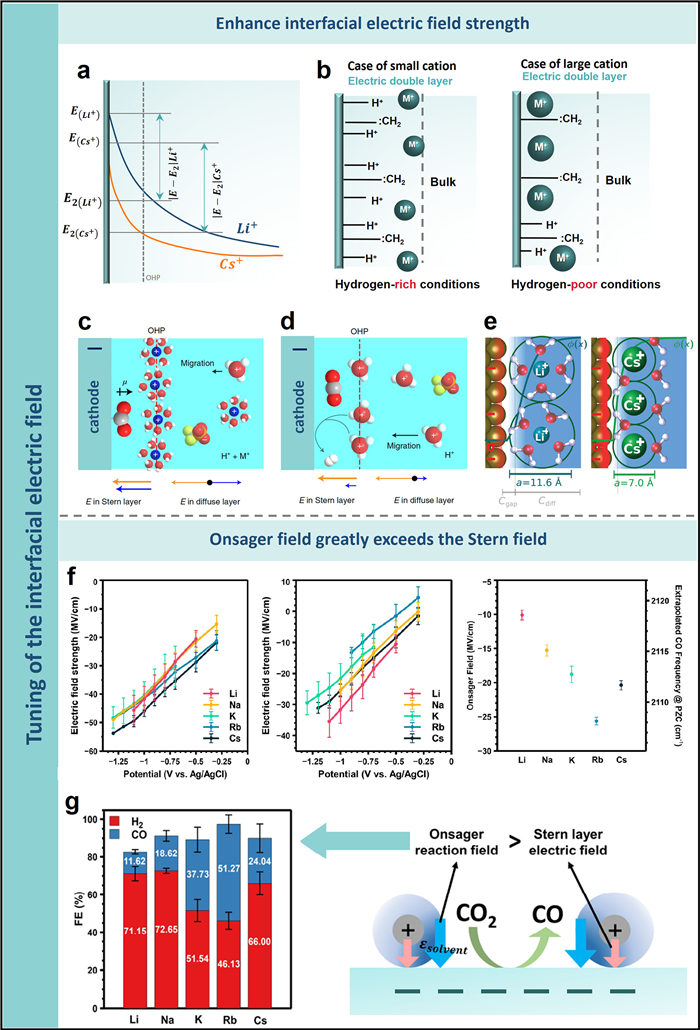
2. Tuning of the interfacial electric fieldThe multifaceted and yet undetermined role of cations in the CO2RR process on negatively charged metal electrodes persistently intrigues researchers. Murata and Hori pioneered a hypothesis elucidating the role of metal cations in this reaction [30]. They postulated that the cathode surface acquires a negative charge during CO2 reduction, triggering cations to adsorb specifically via coulombic forces or covalent bonding. The degree of this specific adsorption is influenced by the cation type, which subsequently alters electrode surface's hydrogen ion density and the OHP potential (Fig. 1a). Smaller radius cations, such as Li+, exhibit high hydration levels and do not readily adsorb to the electrode surface. In contrast, larger cations like K+ and Cs+, possessing smaller hydration shells, demonstrate easier adsorption, initiating a positive potential shift at the OHP (Figs. 1a and b) [30,31]. This shift in OHP potential diminishes the interface's hydrogen ion density, creating a localized high pH environment that promotes the conversion of CO2 to C2+.

|
Download:
|
| Fig. 1. (a) Illustration of potential distribution across the surface of the electrode. Reproduced with permission [30]. Copyright 1991, The Chemical Society of Japan. (b) Conceptual representation of cation species present on the Cu electrode. Reproduced with permission [31]. Copyright 2006, Elsevier. (c) Representation of the EDL near the cathode in trifluoromethanesulfonic acid/metal cation and (d) only trifluoromethanesulfonic acid environments. The spheres in white, red, grey, blue, orange and yellow denote H, O, C, K, S and F atoms, respectively. Reproduced with permission [32]. Copyright 2022, Springer Nature. (e) Visual representation of the root causes of observed cation effects in electrocatalysis influenced by an external field. Reproduced with permission [34]. Copyright 2019, The Royal Society of Chemistry. (f) Combined field strength (left) and electric field intensity of the Stern layer (middle) at different potentials, alongside the estimated CO frequency at the PZC and corresponding Onsager reaction field strength (right). (g) FE for CO and H2 measured at different cations (left) and Illustration of the electric field at the electrode surface (right). Reproduced with permission [36]. Copyright 2022, American Chemical Society. | |
{kind=link}
The electric field at the electrode interface can be boosted through the incorporation of electrolyte cations, thereby fostering the adsorption process of CO2 and mitigating the activation energy requisite for the intermediate formation. To explore the impact of cations on the interfacial electric field, a study by Hu et al. used Au electrode for CO2 electrochemical reduction in an acidic electrolyte. Their findings highlighted that the physically attached hydrated alkali metal cation on the electrode interface triggered a substantial alteration in the electric field arrangement within the EDL [32]. The addition of cations significantly increased the electric field intensity of the stern layer in comparison to the electrolyte without cations, and the electric field strength increased as the negative potential increased. When the electrode charge is below the zero charge point, the cations are attracted by the negatively charged surface, and the electrostatic force causes them to enrich at the OHP. The resulting electric field, combined with the electric field produced via the electrode, further intensifies the field in the stern layer. On the other hand, the electric fields instigated by the cations demonstrate a counteractive trajectory to the electrode-generated electric fields in the diffusion layer. The local electric fields produced by cations shield those generated by the electrodes, inhibiting the migration of hydrogen ions (Fig. 1c). Conversely, in acidic systems lacking cations, only the hydrated hydrogen ion is present at the OHP, where it is reduced to H2 and consumed (Fig. 1d). Consequently, the cation-induced electric field is insufficient to shield the electrode electric field, making the HER reaction more favorable. Furthermore, the study demonstrated that the higher electric field of the stern layer possesses the capacity to engage with the dipole moment of CO2-adsorbed species, stabilizing the associated intermediates and promoting CO2 reduction.
Resasco et al. employed density functional theory (DFT) calculations to establish a correlation between the size of the cations and the intensity of interfacial electric field, demonstrating that larger cations enhance the electric field by accumulating more on the electrode interface. Amplification of the electric field can reduce the *CO2 adsorption energy, facilitating the generation of CO and HCOOH. Moreover, the electrostatic engagement between the electric field and reaction intermediates can lower the overpotential required for the formation of *OCCO or *OCCHO, thus encouraging the production of C2+ compounds [33]. In a similar vein, Chan et al. have constructed a robust theoretical model to interpret the impact of cations. This model was well congruent with experimental data observed on catalysts such as Ag and Cu. The model proposed that cations with diminished hydration shells, like Cs+, encounter the least resistance when adjacent to the electrode. This results in an elevated concentration at the electrode relative to larger cationic hydration shells, such as Li+ (Fig. 1e). The observed concentration disparity induces an enhancement of interfacial charge density and local electric field intensity, thereby fostering the adsorption of CO2 [34].
Recent investigations have proposed that the fluctuations in electric field intensity may not account entirely for the observed reactivity trend associated with cation size. Malkani et al. employed attenuated-total-reflection surface-enhanced infrared spectroscopy (ATR−SEIRAS) spectroscopy on polycrystalline copper films to investigate the activity of alkali cations on CORR, revealing that distinct cations generate a variety of CO absorption sites on the copper surface. A gradual escalation in the ratio of CO adsorbed at the step site versus the terrace site is observed with the progression in cation size from Li+ to K+, reaching equilibrium with greater cations. This trajectory paralleled the Stark tuning rate of CO adsorption as detected across a variety of cationic electrolytes, indicative of the electric field's intensity within EDL. Therefore, it can be inferred that the primary driver for the enhancement in CORR performance from Li+ to K+ is the potency of electric field at the interface. Interestingly, as the cation size extends from K+ to Cs+, the Stark tuning rate stabilizes, while the CORR continues to climb, suggesting the involvement of non-electric field elements in the cationic effect [35]. Subsequent spectroscopic investigations employing organic molecules and cation-chelating agents have illuminated that these factors are influenced by the specific cation and the constitution of the electrochemical interface.
Nevertheless, Baker and collaborators [36] examined the oscillatory Stark shift of CO produced on Au electrodes in an electrolyte containing alkaline metal cation. Their research allowed for the comprehensive evaluation of the total electric field, differentiating its contributions from the Onsager reaction layer and electrochemical Stern field (Fig. 1f). Their findings contrasted with previous research. They proposed that the Stern field had less influence on the kinetics of CO2RR, while the strength of the Onsager reaction layer played a more significant role. Further, they identified a specific relationship between the variation in the strength of the Onsager reaction layer and a trend in CO2RR activity involving different alkali metal cations (Fig. 1g). This correlation suggested that the Onsager layer exerted a more substantial impact than the Stern field, particularly when adsorbed (bending) CO2 was present. However, one outlier in their study was the alkali metal cation Cs+, which consistently showed irregular changes. This irregularity led to further investigation by Smith and his team [37]. Their research concluded that Cs+ caused a smaller decrease in the relative solution tolerance on the OHP surface, primarily due to its less effective polarization of water molecules compared to other alkali metal cations. This result supports Baker's findings. The abnormal electric field strength and CO2RR activity of Cs+ were attributed to its partial solvation in the stern layer, which is in line with the weakening of the strong hydrogen bond water peak in the sum frequency spectrum.
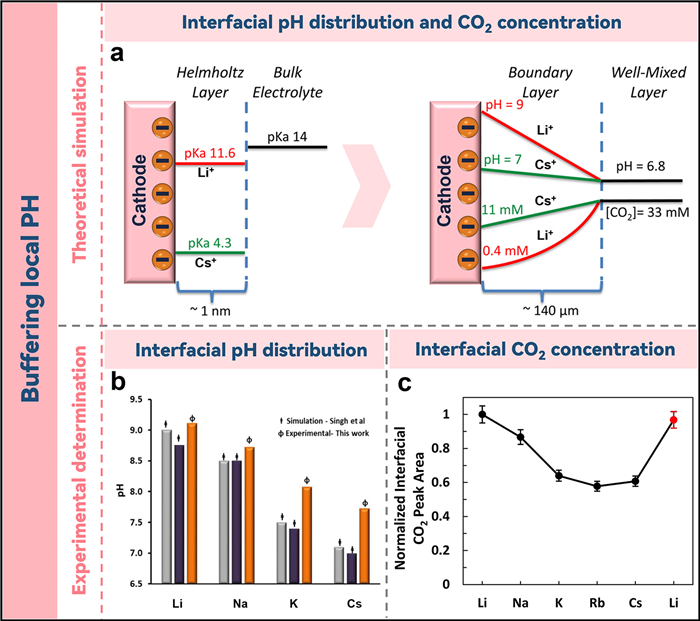
3. Buffering of the local pHThe role of cation species in determining the selectivity of CO2RR products is well established, yet the mechanisms contributing to their overall catalytic efficiency enhancement remain elusive. Bell and colleagues have put forth a conjecture that the hydrolysis of alkaline metal cations in proximity to the cathode surface serves as a pH buffer at the interface, thereby promoting CO2RR whilst concurrently constraining the HER [38]. The buffering capacity is contingent on the pKa value of the interface's hydrated cation, which inversely correlates with cation size. If the pKa of the hydrated cation falls below the interfacial pH, protons are liberated from the dissociation of the hydrated water, thereby maintaining the stability of the local pH. Hydrated cation's pKa values were observed to align in the order Li+ > Na+ > K+ > Cs+. Computational analysis indicated a cathode pH decline from 9 to 7 alongside an increase in cation size from Li+ to Cs+ (Fig. 2a), implying a superior buffering capacity for larger cations such as Cs+ that bolsters CO2RR. Furthermore, an approximately 30-fold augmentation (from 0.4 mmol/L to 11 mmol/L) in cathode CO2 concentration was noted with the enhanced cation dimension. The minimum cathode CO2 concentration was recorded in the presence of Li+, as an elevated cathode pH (> 8.5) promotes the transformation of CO2 to CO32− and HCO3−. Polarization loss is induced by increased cathode pH and a reduction in CO2 local density. However, larger cations with a more potent buffering capacity and higher cathodic CO2 concentration significantly enhance CO2RR overall performance. This premise was further substantiated by Zhang et al., who utilized a rotating ring-disc electrode (RRDE) assembly to monitor alterations in interfacial pH during the aqueous CO2RR [39].

|
Download:
|
| Fig. 2. (a) Hydration of Li+ and Cs+ in the Helmholtz layer and bulk electrolyte, along with the pH and CO2 concentration distribution in the EDL. Reproduced with permission [38]. Copyright 2016, American Chemical Society. (b) pH equilibrium at the metal-electrolyte interface in M2CO3 solutions (M = Cs, K, Na, Li). Reproduced with permission [40]. Copyright 2017, American Chemical Society. (c) Monitoring local CO2 density. Reproduced with permission [41]. Copyright 2020, American Chemical Society. | |
{kind=link}
Contrarily, Cuesta et al. observed an anomaly when they utilized ATR−SEIRAS to scrutinize the interface pH [40]. Based on the simulation and experimental results, they found that although the buffering capacity still increased with cation size, Cs+ failed to maintain the interface pH as expected (~7), suggesting that the pH buffering effect of cationic hydrolysis concluded by Bell et al. might have been overestimated (Fig. 2b). The researchers associated this incongruity in buffering effect to an overestimated surface charge density of the electrode, which consequently led to an exaggerated deduction of the pKa reduction in cationic hydrolysis. Interestingly, when electrolytes were formulated using salts of lower purity, the behavior pattern of alkaline cations exhibited an inverse buffering effect. This highlights the importance of careful chemical selection in CO2RR studies, given the considerable impact of electrolyte purity on the observed trends.
In a subsequent investigation, Malkani et al. scrutinized the effect of cations on CO2RR by tracking alterations in interfacial CO2 concentration through the monitoring of the stretch vibration peak at 2343 cm−1. The findings suggested that throughout the CO2RR procedure on the Au electrode, a decrease in the local CO2 concentration was noticed, which corresponded to an enlargement of cation size (Fig. 2c). The result challenges the buffering hypothesis put forth by Bell et al., which is related to interfacial cations. Instead of the earlier proposed buffering effect, Malkani et al. ascribed the changes in interfacial CO2 concentration to the speed of CO2 consumption, facilitated either by CO2RR or by interaction with electrode-produced OH− [41].
Modifications of localized pH on the catalyst surface can be broadly segmented into two domains: the buffering effects, and OH− depletion by CO2. This underscores the necessity for additional investigations to probe the impact of cations at high current densities pertinent to industrial applications. Such inquiries are not merely substantial but also contribute to the clarification of the previously posed query. This is especially critical given that, at these elevated current intensities, the cationic buffering effect markedly surpasses its threshold.
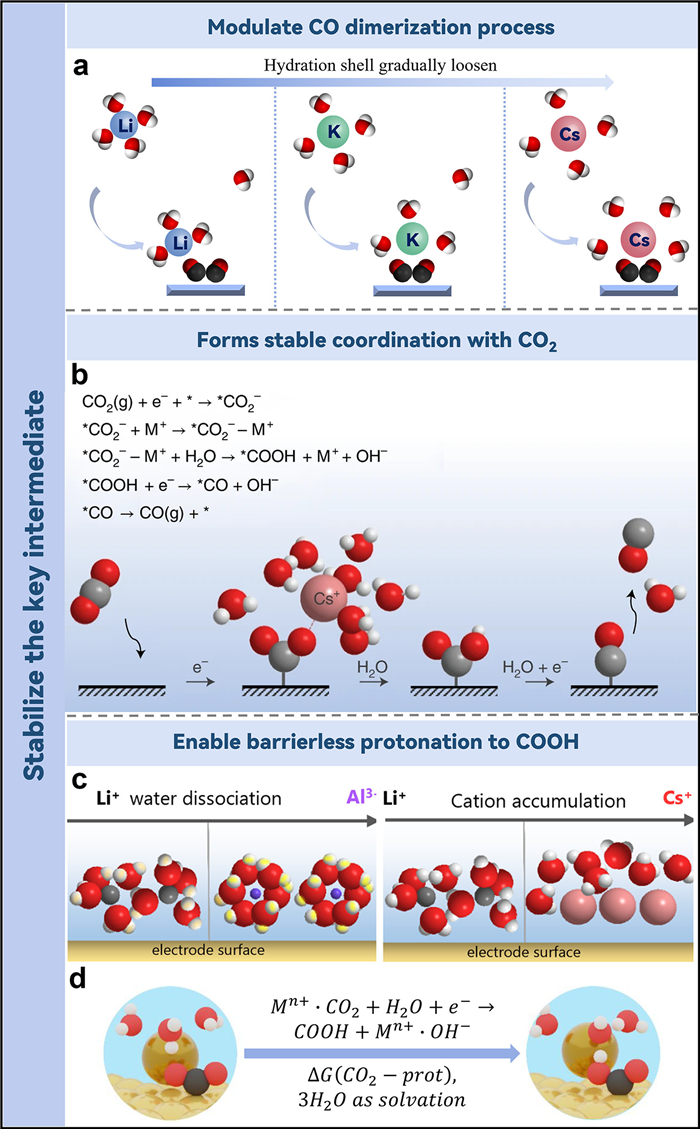
4. Stabilization of the key intermediatesWhile the buffering of local pH or adjustment of the interfacial electric field can elucidate the enhanced efficiency of CO2 reduction, these theories inadequately address the observed phenomenon that CO2RR scarcely transpires in an electrolyte devoid of metal cations. In addition to the aforementioned mechanisms, cations have also been proposed to directly participate in the promotion of CO2RR by interacting with relevant intermediates. Akhade et al. employed DFT to scrutinize the influence of specifically adsorbed ions on the binding strength of intermediates on various crystal facets of Cu [42]. At overpotentials for CO2RR, the computations suggested a thermodynamic advantage in the weak coverage of particularly attached cations. Upon the adsorption of K+, a significant diminution in the activation energy barriers for the transformation of CO* to CHO* is observed. This can be theoretically explicated by the augmented interface dipole moment ensuing from the enhanced charge retention of K*. This circumstance expedites the electron transference from the interface to the transition state. Inversely, the impediment for the CO* to COH* reduction escalates by 0.70 eV in the presence of adsorbed K+, attributable to the destabilizing influence of K+ on the COH* formation transition state. Their work suggests that specifically adsorbed cations enhance the stability of CHO* and CO*, and promote the formation of C2 products. Additionally, Yang et al. utilized the method of theoretical simulation to explore the role of cation-containing explicit solvents on CO dimerization in the OCCO process [43]. The experimental outcomes demonstrated that the reaction-free energy barrier of C—C coupling gradually decreased as cation size increased from Li+ to Cs+, suggesting that larger cations have a stronger propensity to promote CO—CO coupling and produce the multiple carbon products. The researchers concluded that the hydrated cation coordinates with O in the OCCO intermediate by shedding H2O molecules from its hydration shell after analyzing the coordination environment between cations and OCCO intermediates (Fig. 3a). In contrast to the hydrated shells of Li+, the shells of K+ and Cs+ are relatively loose and will coordinate with the oxygens in OCCO to form a stable cation-intermediate complex.

|
Download:
|
| Fig. 3. (a) Schematic representation of the dynamic coordination of the alkali metal cations Li, K and Cs with H2O and reaction intermediates, utilizing color-coding (H: white, O: red, C: black). Reproduced with permission [43]. Copyright 2021, American Chemical Society. (b) Diagrammatic portrayal of the cation's interaction with the negatively charged *CO2– intermediate. Reproduced with permission [44]. Copyright 2021, Springer Nature. (c) Diagrammatic depiction of the engagement of varying cation species with the electrode surface. (d) Conceptual model for the initial and terminal state of CO2 protonation. Reproduced with permission [45]. Copyright 2022, American Chemical Society. | |
{kind=link}
The aforementioned analysis is predicated on theoretical simulations. The research work of Koper and associates provides robust experimental and theoretical evidence underscoring the pivotal role of cations in CO2 electrocatalysis, particularly as stabilizers of reaction intermediates [44]. By leveraging scanning electrochemical microscopy (SECM), the researchers discovered that without metal cations in the electrolyte, the CO2RR ceases to occur. Their DFT unveiled that partially desolvated cations assist in CO2RR via three promotional mechanisms. Primarily, these cations provide more effective stabilization of CO2 compared to water molecule solvation alone, facilitated by short-range M+−O(CO2) electrostatic engagement and medium-range interactions between *CO2− and an electric field generated by cations. In the absence of cations, this stabilization is diminished and insufficient to instigate CO2 reduction. Secondly, the presence of cations decreases the O—C—O linear molecule angle from 180° to less than 140°, thus activating CO2. Finally, cations enhance electron transfer from the interface of catalyst to CO2, a crucial step for the generation of the *CO2− intermediate, which is the rate-determining step in CO2RR. They noted that the behavior variations among alkaline cations for CO2RR are associated with their distinct concentrations at the OHP and their unique capacities to engage with negatively charged adsorbates. In comparison to Li, alkaline cations such as Na+, K+, and Cs+, which exhibit a less rigid hydration shells, demonstrate an increased propensity to accumulate in the OHP. This accumulation serves to stabilize *CO2− through the M+−O(CO2) interactions. As a result, the study proposed that cations play a pivotal role as a promoter in the reduction process, forming a complex with CO2 to aid the formation of the *CO2− intermediate (Fig. 3b).
They further examined the effects of mono- and multivalent metal cations on the HER and CO2RR on gold electrodes [45]. The study highlighted the significant role of acidic cations' promotional effects on water reduction in influencing CO2RR and HER performance. The stability of the *CO2− intermediate and the concentration of cations at the OHP were identified as critical elements in this process. The highest water reduction activity was found for weakly hydrated trivalent species, particularly Nd3+ and Ce3+. Despite the greater acidity of cations with smaller ionic radii, their high solvation energy limits their aggregation in proximity to the interface (Figs. 3c and d). Trivalent cations were found to promote both *CO2− stabilization and water dissociation. In contrast, Cs+ and Ba2+ were associated with high CO2 reduction activity at high overpotentials due to their ability to stabilize the *CO2− intermediate, and demonstrating poor kinetics for water dissociation.
Besides, alkyl ammonium cations are also believed to stabilize the related intermediates involved in CO2RR. Sun et al. constructed an artificial electrode/electrolyte to alter the CO2 reduction reaction pathway through enveloping quaternary ammonium cationic (R4N+) surfactants on the electrode surface [46]. The results of their experiments revealed that linear straight-chain cationic surfactants can promote the formation of HCOOH, and their selectivity increases with the length of the carbon chain (TMAC < OTAC < DTAC < CTAC). While quaternary ammonium cationic surfactants with different branched chains have a strong selectivity for CO, and their activity increases with the length and complexity of the side chain, particularly those with benzene rings (BTEAC > TTAC > BTMAC > TBAC > TEAC). The calculation results show that straight-chain R4N+ can stabilize the *OCHO intermediate, thereby driving CO2 reduction to formic acid. While increasing the side chain length or introducing a benzene ring into the branched chain reduces the affinity for *CO species, leading to the repulsion of *CO species and enabling CO desorption. This strategy is also applicable to electrodes other than Cu such as Sn and Zn.
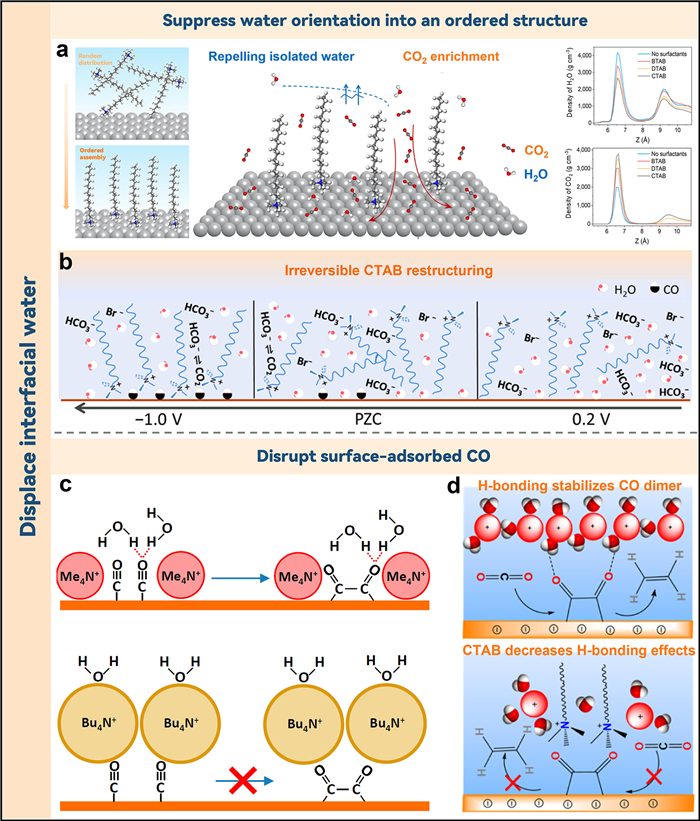
5. Regulation of the interfacial waterLong-chain organic cations with hydrophobic properties have been postulated to affect the hydrolysis reaction rate by disrupting the structure of interfacial water. To elucidate this effect of long-chain organic cation on CO2RR, Li and associates employed in situ ATR-SEIRAS investigations to examine the catalytic characteristics of commercial silver electrodes within the systems of CO2 and H2O electrocatalytic co-reduction [47]. The cationic surfactants modulated the interface water environment by repulsing the interface and inhibiting the orientation of water to form an ordered structure, which stimulates CO2 enrichment at charged interfaces. With an increase in bias potential, cetyltrimethylammonium bromide (CTAB) could effectively inhibit the approach of hydrogen atoms in interfacial water to the electrode surface. This inhibition was ascribed to the long Ag-H distance in the CTAB-contained system, which diminished hydrolysis activity. Long-chain surfactants form an ordered assembly structure, creating a gas-hydrophobic channel that boosts gas mass transfer proficiency and catalytic selectivity, while diminishing the HER from electrolytic water. The molecular dynamics simulation confirmed the experimental findings that quaternary ammonium surfactants could decrease the water dissociation activity and increase the generation of CO products. Eventually, they observed that hydrophobicity increased with the length of the alkyl line, resulting in enhanced CO2 adsorption ability and higher CO2 reduction activity, with a trend of CTAB > DTAB > OTAB > BTAB (Fig. 4a). Hall et al. reported the occurrence of a comparable phenomenon in their study, wherein they employed SEIRAS to investigate the kinetics of the interfacial water environment with CTAB acting as a molecular promoter [48]. The accumulation and irreversible reorientation of CTAB on the electrode surface caused a perturbation in the binding configuration of CO and destroyed the hydrogen bond network of water (Fig. 4b). As a consequence, the rate of CO2RR increased by a factor of ten and the HER rate decreased four-fold.

|
Download:
|
| Fig. 4. (a) The surfactants located at the electrically charged interface transition from a disordered to an almost ordered assembly as the potential increases (right). The middle image provides a diagrammatic representation of the surfactant arrangement at this charged interface, while the left image depicts density distributions of H2O and CO2, computed using molecular dynamics for various surfactants. Reproduced with permission [47]. Copyright 2022, American Chemical Society. (b) The effect of CTAB's potential-dependent adsorption on interfacial water concentration during cathodic scan is depicted. Reproduced with permission [48]. Copyright 2020, American Chemical Society. (c) Sluggish formation of CO dimers due to the displacement of interfacial water by these hydrophobic cations. Reproduced with authorization [49]. Copyright 2020, American Academy of Sciences. (d) A mechanism suggesting the prevention of C—C coupling with CTAB is presented. Reproduced with permission [50]. Copyright 2020, American Chemical Society. | |
{kind=link}
Additionally, Waegele et al. applied differential electrochemical mass spectrometry (DEMS) to evaluate CO reduction products in the electrolyte containing different quaternary alkylammonium cations (methyl4N+, ethyl4N+, propyl4N+ and butyl4N+) [49]. The ethylene was generated in the electrolytes containing methyl4N+ and ethyl4N+ cations but was not detected in the electrolytes in propyl4N+ and butyl4N+. Further SEIRAS spectroscopy analysis revealed these alkylammonium cations were weakly affected by the interfacial electric field and would not impede the availability of the carbon monoxide adsorption sites. The existence of significant amounts of propyl4N+ and butyl4N+, which interfered with the intermolecular interactions between interfacial water and surface-adsorbed CO, has been suggested as the cause of the variation in ethylene selectivity. Their large spatial site resistance and high hydrophobicity repelled interfacial water from contact with the adsorbed CO, preventing it from forming hydrogen bonds with the terminal oxygen to stabilize the process of CO dimerization (Fig. 4c).
The effect of the coexistence of alkaline metal cations and CTAB surfactant on CO2RR was also investigated. It was found that the efficiency and selectivity of the CO2RR, when executed in the presence of CTAB, appear to be largely unaffected by the specific properties of the alkaline metal cations involved. The limited availability of electrolyte cations on the electrode surface, due to competition between CTAB molecules and alkali metal cations for positions on the OHP, is the reason for this observation. However, Cs+ exhibited a strong competitive absorption against CTAB and accumulated more on the surface, resulting in a high activity of HCOO− (Fig. 4d top) in electrolytes containing both CTAB and Cs+. In this case, the hydrophobic effect at the interface spurred forth by the buildup of CTAB nevertheless prevented the formation of ethylene. The CTAB displaced the interfacial water and destroyed its hydrogen bond structure, thereby hindering the diffusion of local proton sources and impeding the process of CO2 dimerization to generate ethylene (Fig. 4d down) [50].
6. Summary and outlookThis comprehensive critique delivers an extensive exploration of metal cations' role in the CO2RR. By diligently dissecting numerous theoretical and experimental studies, we illuminate the significant influence of cations on the activity, selectivity, and operational mechanism of the CO2RR. Our examination pinpoints four principal strategies wherein cations modulate the CO2RR, incorporating an array of interlinked procedures, which encompass:
(1) Cations are believed to adsorb to the cathode surface during CO2 reduction, with the degree of adsorption influenced by the cation type. Larger cations like K+ and Cs+ are found to adsorb more readily due to smaller hydration shells, inducing a positive potential shift at the OHP. This shift fosters a localized high pH environment, facilitating the reduction of CO2 to C2+. Additionally, the presence of electrolyte cations can boost the electric field at the electrode interface, promoting the adsorption of CO2 and reducing the activation energy of intermediate formation. The size of the cation has been correlated with the interfacial electric field strength, with larger cations enhancing the field by accumulating more on the electrode interface. Nevertheless, contemporary research suggest that fluctuations in electric field intensity may not entirely account for the observed reactivity trend associated with cation size. Other factors, such as the distribution of CO adsorption sites on Cu and the influence of the cationic identity on both electric and non-electric field components, also play a crucial role.
(2) Cations exert a substantial impact on the selectivity of CO2RR products, whereby larger cations, notably Cs+, exhibit enhanced buffering capacity that promotes CO2RR. This phenomenon arises from the hydrolysis of alkaline metal cations in proximity to the cathode surface, which serves as a pH buffer. While certain investigations propose that larger cations augment the concentration of CO2 at the cathode, thereby bolstering the overall performance of CO2RR. Other studies question this conclusion. They propose that changes in the interfacial CO2 concentration originate from the rate of CO2 consumption, facilitated by CO2RR or react with OH− species.
(3) Cations assume a pivotal function by engaging with reaction intermediates, endorsing specific pathways, and providing stability to CO2. For example, the adsorption of K+ ions mitigates energy barriers, thereby augmenting the reduction of CO* to CHO*. Cations of larger sizes facilitate C—C coupling and the production of C2+ products. They also contribute to the stabilization of CO2 via M+−O(CO2) and subsequently expedite electron transfer to form *CO2− intermediate. The presence of cations, particularly those with less rigid hydration shells (Cs+ and K+), is indispensable for the progression of CO2 reduction. Moreover, alkyl ammonium cations and cationic surfactants can stabilize related intermediates and modulate the CO2 reduction pathway.
(4) Long-chain organic cations are thought to displace interfacial water and modify the hydrolysis reaction rate, thereby distorting the binding configuration of CO and shattering the hydrogen bond network of water. These effects result in an elevated rate of CO2RR and a reduced rate of HER. Alkylammonium cations reduced the selectivity of ethylene by impeding interfacial water from touching down with adsorbed CO. Quaternary ammonium surfactants reduce the dissociation activity of water while increasing the ability of CO2 adsorption and CO2 reduction activity, with CTAB demonstrating the highest level of activity.
Despite the effectiveness of current models in rationalizing observed trends in various experiments, their generalizability remains unproven. The investigation of the electrolyte cation-mediated mechanism in the electrocatalytic carbon dioxide process is a complex task, primarily due to the intricate nature of the system. The impact of cations on electrocatalytic performance is governed by a multitude of factors, including charge density, cation sizes, types, and the extent of solvation. The cation-induced mechanism is characterized by sophisticated interconnections between the electrolyte and the electrode interface. These interactions are often marked by reciprocal limitations or cooperative synergies, thus creating a complex dynamic that is not easily deconstructed or understood. This multifaceted complexity significantly elevates the level of intricacy involved in experimental scrutiny. Consequently, this poses substantial challenges to researchers attempting to isolate and analyze individual components or effects within the system. In situ methodologies, such as SEIRAS and surface-enhanced Raman spectroscopy (SERS), have been recognized as pivotal in unmasking the molecular-level implications of electrolyte cations, particularly in relation to local pH, interfacial electric field, and the structure of interface water. However, these in situ techniques inherently fall short in scrutinizing environmental interfacial modifications that transpire over smaller coverage areas, thereby underscoring the necessity for more sophisticated spectroscopic methodologies. Approaches such as DFT, ab initio Born-Oppenheimer molecular dynamics simulations (AIMD), and free energy sampling have been instrumental in investigating the effects of electrolyte cations on absorption and pivotal intermediates, and ought to be considered as adjuncts to spectroscopy.
Importantly, the absence of uniform protocols for evaluating the repercussions of cation-mediated mechanisms on the CO2RR process can engender discrepancies across disparate research entities, thereby obstructing comparative analyses and generalization of findings. To attain a more comprehensive understanding of the role of cations in interfacial reactions, the employment of avant-garde techniques such as computational analysis, isotope tracking, and in situ mass spectrometry is advocated. These cutting-edge methodologies offer the potential to yield deeper insights into the complex mechanisms that underlie cation-mediated electrolytes and expedite further innovations in the realm of electrochemical CO2 reduction. This formidable task mandates interdisciplinary expertise encompassing electrochemistry, materials science, and computational modeling to achieve an all-encompassing grasp of the subject matter.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support from National Natural Science Foundation of China (Nos. 22109099 and 22072101), "Chen Guang" Project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation (No. 21CGA66), Natural Science Foundation of Jiangsu Province (No. BK20211306) and Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions is gratefully acknowledged. We also thank Soochow Municipal laboratory for low carbon technologies and industries.
| [1] |
S. Jin, Z.M. Hao, K. Zhang, et al., Angew. Chem. Int. Ed. 60 (2021) 20627-20648. DOI:10.1002/anie.202101818 |
| [2] |
G. Wu, S. Guo, Energy Lab 1 (2023) 230001. |
| [3] |
J. Rogelj, M.D. Elzen, N. Höhne, et al., Nature 534 (2016) 631-639. DOI:10.1038/nature18307 |
| [4] |
P.D. Luna, C. Hahn, D. Higgins, et al., Science 364 (2019) 364. |
| [5] |
L.K. Xiong, X. Zhang, L. Chen, et al., Adv. Mater. 33 (2021) 2101741. DOI:10.1002/adma.202101741 |
| [6] |
Y. Gao, Qian Wu, X.Z. Liang, et al., Adv. Sci. 7 (2020) 1902820. DOI:10.1002/advs.201902820 |
| [7] |
Z. Chen, T. Wang, B. Liu, et al., J. Am. Chem. Soc. 142 (2020) 6878-6883. DOI:10.1021/jacs.0c00971 |
| [8] |
G. Gregorio, T. Burdyny, A. Loiudice, et al., ACS Catal. 10 (2020) 4854-4862. DOI:10.1021/acscatal.0c00297 |
| [9] |
Y. Wang, P. Wang, X. Lv, et al., Joule 2 (2018) 2551-2582. DOI:10.1016/j.joule.2018.09.021 |
| [10] |
B. Zhang, J. Zhang, M. Hua, et al., J. Am. Chem. Soc. 142 (2020) 13606-13613. DOI:10.1021/jacs.0c06420 |
| [11] |
P.D. Luna, R. Quintero-Bermudez, C.T. Dinh, et al., Nat. Catal. 1 (2018) 103-110. DOI:10.1038/s41929-017-0018-9 |
| [12] |
L.K. Xiong, X. Zhang, H. Yuan, et al., Angew. Chem. Int. Ed. 60 (2021) 2508-2518. DOI:10.1002/anie.202012631 |
| [13] |
Z.Z. Wu, F.Y. Gao, M.R. Gao, Energy Environ. Sci. 14 (2021) 1121-1139. DOI:10.1039/D0EE02747B |
| [14] |
L.K. Xiong, Z. Sun, X. Zhang, et al., Nat. Commun. 10 (2019) 3782. DOI:10.1038/s41467-019-11766-w |
| [15] |
T. Gao, A. Kumar, Z. Shang, et al., Chin. Chem. Lett. 30 (2019) 2274-2278. DOI:10.1016/j.cclet.2019.07.028 |
| [16] |
Q. Zheng, C. Chen, S. Cao, et al., Chin. Chem. Lett. 34 (2023) 107273. DOI:10.1016/j.cclet.2022.02.078 |
| [17] |
X. Chen, J. Chen, N.M. Alghoraibi, et al., Nat. Catal. 4 (2020) 20-27. DOI:10.1038/s41929-020-00547-0 |
| [18] |
F. Li, A. Thevenon, A.R. Hernández, et al., Nature 577 (2020) 509-513. DOI:10.1038/s41586-019-1782-2 |
| [19] |
Z. Xin, Z. Yuan, J. Liu, et al., Chin. Chem. Lett. 34 (2023) 107458. DOI:10.1016/j.cclet.2022.04.056 |
| [20] |
H. Shen, C. Han, B. Wang, et al., Chin. Chem. Lett. 33 (2022) 3721-3725. DOI:10.1016/j.cclet.2021.10.063 |
| [21] |
H. Yu, X. Wu, Q. Mu, et al., Chin. Chem. Lett. 32 (2021) 3833-3836. DOI:10.1016/j.cclet.2021.05.035 |
| [22] |
F. Hao, Y. Wang, Z. Fan, Energy Lab 1 (2023) 220023. |
| [23] |
X. Shi, L. Cao, M. Chen, et al., Chin. Chem. Lett. 33 (2022) 5023-5029. DOI:10.1016/j.cclet.2022.01.066 |
| [24] |
Y.J. Sa, C.W. Lee, S.Y. Lee, et al., Chem. Soc. Rev. 49 (2020) 6632-6665. DOI:10.1039/D0CS00030B |
| [25] |
W. Paik, T.N. Andersen, H. Eyring, Electrochim. Acta 14 (1969) 1217-1232. DOI:10.1016/0013-4686(69)87019-2 |
| [26] |
Y. Qiao, W. Lai, K. Huang, et al., ACS Catal. 12 (2022) 2357-2364. DOI:10.1021/acscatal.1c05135 |
| [27] |
Y. Lum, B. Yue, P. Lobaccaro, et al., J. Phys. Chem. C 121 (2017) 14191-14203. DOI:10.1021/acs.jpcc.7b03673 |
| [28] |
A. Schizodimou, G. Kyriacou, Electrochim. Acta 78 (2012) 171-176. DOI:10.1016/j.electacta.2012.05.118 |
| [29] |
J.E. Huang, F. Li, A. Ozden, et al., Science 372 (2021) 1074-1078. DOI:10.1126/science.abg6582 |
| [30] |
A. Murata, Y. Hori, Bull. Chem. Soc. Jpn. 64 (1991) 123-127. DOI:10.1246/bcsj.64.123 |
| [31] |
S. Kaneco, H. Katsumata, T. Suzuki, K. Ohta, Electrochim. Acta 51 (2006) 3316-3321. DOI:10.1016/j.electacta.2005.09.025 |
| [32] |
J. Gu, S. Liu, W. Ni, et al., Nat. Catal. 5 (2022) 268-276. DOI:10.1038/s41929-022-00761-y |
| [33] |
J. Resasco, L.D. Chen, E. Clark, et al., J. Am. Chem. Soc. 139 (2017) 11277-11287. DOI:10.1021/jacs.7b06765 |
| [34] |
S. Ringe, E.L. Clark, J. Resasco, et al., Energy Environ. Sci. 12 (2019) 3001-3014. DOI:10.1039/C9EE01341E |
| [35] |
A.S. Malkani, J. Li, N.J. Oliveira, et al., Sci. Adv. 6 (2020) eabd2569. DOI:10.1126/sciadv.abd2569 |
| [36] |
Q. Zhu, S.K. Wallentine, et al., JACS Au 2 (2022) 472-482. DOI:10.1021/jacsau.1c00512 |
| [37] |
D. Bohra, J.H. Chaudhry, T. Burdyny, et al., Energy Environ. Sci. 12 (2019) 3380-3389. DOI:10.1039/C9EE02485A |
| [38] |
M.R. Singh, Y. Kwon, Y. Lum, et al., J. Am. Chem. Soc. 138 (2016) 13006-13012. DOI:10.1021/jacs.6b07612 |
| [39] |
F. Zhang, P.A.C. Co, Angew. Chem. Int. Ed. 59 (2020) 1674-1681. DOI:10.1002/anie.201912637 |
| [40] |
O. Ayemoba, A. Cuesta, ACS Appl. Mater. Interfaces 9 (2017) 27377-27382. DOI:10.1021/acsami.7b07351 |
| [41] |
A.S. Malkani, J. Anibal, B. Xu, ACS Catal. 10 (2020) 14871-14876. DOI:10.1021/acscatal.0c03553 |
| [42] |
S.A. Akhade, I.T. McCrum, M.J. Janik, J. Electrochem. Soc. 163 (2016) F477-F484. DOI:10.1149/2.0581606jes |
| [43] |
H. Liu, J. Liu, B. Yang, ACS Catal. 11 (2021) 12336-12343. DOI:10.1021/acscatal.1c01072 |
| [44] |
M.C.O. Monteiro, F. Dattila, B. Hagedoorn, et al., Nat. Catal. 4 (2021) 654-662. DOI:10.1038/s41929-021-00655-5 |
| [45] |
M.C.O. Monteiro, F. Dattila, N.L. ópez, M.T.M. Koper, J. Am. Chem. Soc. 144 (2022) 1589-1602. DOI:10.1021/jacs.1c10171 |
| [46] |
Y. Zhong, Y. Xu, J. Ma, et al., Angew. Chem. Int. Ed. 59 (2020) 19095-19101. DOI:10.1002/anie.202005522 |
| [47] |
W. Ge, Y. Chen, Y. Fan, et al., J. Am. Chem. Soc. 144 (2022) 6613-6622. DOI:10.1021/jacs.2c02486 |
| [48] |
Z.Q. Zhang, S. Banerjee, V.S. Thoi, A.S. Hall, J. Phys. Chem. Lett. 11 (2020) 5457-5463. DOI:10.1021/acs.jpclett.0c01334 |
| [49] |
J. Li, X. Li, C.M. Gunathungea, M.M. Waegelea, Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 9220-9229. DOI:10.1073/pnas.1900761116 |
| [50] |
S. Banerjee, Z.Q. Zhang, A.S. Hall, V.S. Thoi, ACS Catal. 10 (2020) 9907-9914. DOI:10.1021/acscatal.0c02387 |