 2024, Vol. 35
2024, Vol. 35 


b College of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang 330022, China;
c Engineering Center of Catalysis and Synthesis for Chiral Molecules, Fudan University, Shanghai 200433, China;
d Shanghai Engineering Center of Industrial Catalysis for Chiral Drugs, Shanghai 200433, China
Five-membered nitrogen-containing heterocycles with quaternary stereocenters are among the most coveted scaffolds in natural products and pharmaceutical agents (Scheme 1A) [1,2]. In particular, chiral spirocyclic frameworks derived from such heterocycles are found in many bioactive molecules, which feature intricate three-dimensional structures and increased synthetic difficulty [3-5]. Over the past decades, Pd-catalyzed annulation reactions based on zwitterionic π-allyl palladium complexes have become a powerful method for the asymmetric construction of spirocyclic molecules (Scheme 1B) [6-9]. For example, Trost demonstrated the cycloaddition of azlactone alkylidenes with vinylcyclopropanes, which could be deprotected to yield the corresponding amino acid derivatives [10]. Guo achieved the [4 + 2] cycloaddition of 2-methylidenetrimethylene carbonate with pyrazolone-based alkenes, delivering the chiral spiro-pyrazolones in good stereoselectivity [11]. Liu developed cycloaddition of vinyl cyclopropanes with α,β-unsaturated imines in situ generated from aryl sulfonyl indoles [12]. More recently, Fu and Chen independently reported the synthesis of spirooxindoles using asymmetric annulation strategies (Scheme 1C) [13,14].

|
Download:
|
| Scheme 1. Research backgrounds and this study. | |
On the other hand, adaptations of such chemistry to the synthesis of spiro-pyrrolidine-2,3-diones and other pyrrolidine-based spirocyclic molecules remain elusive [15]. Such nitrogen heterocycles also serve as a versatile precursor to β2,2-amino acids upon oxidation (Scheme 1D) [16]. Dissection of the spirocyclic framework reveals that the annulation with the dioxopyrrolidine-derived alkene 1 is a promising synthetic strategy to approach the counterpart. In this context, alkene 1a is frequently deployed as an electrophilic counterpart for constructing chiral pyrrolidine heterocycles [17,18]. However, the installation of a quaternary carbon chiral center from 1a has only one example [19]. Derivatization of 1a is usually initiated by the conjugate addition of a nucleophile to generate intermediate A. The ambident reactivity of this intermediate has posed difficulty in controlling the chemoselectivity because O-attack is often more favorable than C-attack of the crowded enol carbon atom of Int-A (Scheme 1E) [20-22]. In addition, issues of stereoselectivity can arise from distinguishing the enantiotopic faces of dioxopyrrolidines and the free rotation of the pendant C—C bond of the intermediate.
We envisioned that the C—C bond forming process at the tertiary carbon of A could be implemented by Pd-catalyzed allylation. The decarboxylation of 2-methylidenetrimethylene carbonate 2a generated the dipolarophile that underwent [4 + 2] cycloaddition to furnish tetrahydropyran motifs. Herein, we reported a palladium-catalyzed asymmetric [4 + 2] annulation of 1a with 2-methylidenetrimethylene carbonate 2a, which providing a convergent access to chiral tetrahydropyran-fused spiro-pyrrolidine-2,3-diones containing adjacent quaternary and tertiary stereocenters (Scheme 1F).
We began our investigations with the dioxopyrrolidine derivate 1a and carbonate 2a. The Pd-catalyzed annulation reaction was performed in toluene at 25 ℃ in the presence of Mg(OEt)2 (Table 1). The combination of Pd2(dba)3‧CHCl3 and substituted Segphos scaffolds was investigated at first (Table 1, entries 1–3), and the L3 was the privileged ligand, leading to the formation of the spirocyclic product 3a in high yields (88%) and excellent stereoselectivity (>20:1 dr, 99% ee) after 4 h (Table 1, entry 3). In contrast, lowered yields and enantioselectivity were observed for the reactions with sterically less hindered ligands Segphos L1 and DM-Segphos L2 (Table 1, entries 1 and 2). Other chiral phosphine ligands were also assessed for the reaction. Moderate enantioselectivities were yielded with C2-symmetric diphosphines, as exemplified by L4 and L5 (Table 1, entries 4 and 5). Other tested ligands (L6-L8) were also presented low efficiency.
|
|
Table 1 Optimization of ligands.a |

We highlighted additional results that were noteworthy in Table 2. Variation of Pd catalyst from Pd2(dba)3‧CHCl3 to other Pd sources resulted in reduced enantioselectivity (Table 2, entries 2 and 3). The effect of additives was shown in entries 4 and 5. Utilization of other Lewis acids, including Al(Oi-Pr)3 and Ti(Oi-Pr)4, afforded 3a with good enantioselectivity, albeit with compromised yields (69% and 43%). Furthermore, only Pd2(dba)3‧CHCl3 and Mg(OEt)2 did not promote the reaction (Table 2, entry 6), and the reaction without Mg(OEt)2 led to a decline in both yield and stereoselectivity (Table 2, entry 7). A solvent examination revealed that toluene was optimal (Table 2, entries 8 and 9), while employing CH2Cl2 as the solvent resulted in low yield and enantioselectivity (Table 2, entry 9).
|
|
Table 2 Screen of reaction conditions.a |

{kind=link}
With the optimized conditions established, we investigated the scope of dioxopyrrolidine derivatives (1). As presented in Scheme 2, aryl groups possessing diverse substitution patterns were compatible with the current method. The electronic effect of the vinyl aromatic ring was evaluated using a series of para-substituents (3b-3l). Specifically, the substrates bearing both the electron-rich (3i) and electron-poor groups (3j-3l) were well-tolerated, providing the desired spiro-pyrrolidines in high yields and exceptional enantioselectivities. The analogues bearing substitutions at the meta- (3m-3o) and ortho-positions (3q-3s) of the phenyl rings also demonstrated good yield and excellent ee value. We also obtained the spirocyclic products containing pendant thiophenyl (3t) and cyclohexyl (3u) groups. The reduced enantioselectivity of 3u (38% ee) suggested that π bond of the aromatic rings on enones might have a unique effect on stereoselectivity. In addition, pyrrolidines with different N-substituents were also suitable substrates. For example, 3v and 3w containing extended carbon chains were formed in satisfactory yields and excellent enantioselectivities. Upon examining the electronic properties of the phenyl ring of the N-benzyl groups, the desired reactions proceeded well to furnish 3x-3ac in generally good yields with high enantiomeric excesses. We sought to expand the limitations of the reaction by incorporating 2b as a decarboxylation partner. The annulation reaction occurred at an elevated reaction temperature of 60 ℃, affording the corresponding product 3a with good stereoselectivities, but the yield was reduced to 18% (Scheme 3).

|
Download:
|
| Scheme 2. Substrate scope for the [4 + 2] cycloaddition of enones 1 and 2a. Reaction conditions: reactions were performed with 1a (0.1 mmol), 2a (0.15 mmol), Pd2(dba)3‧CHCl3 (5 mol%), Mg(OEt)2 (15 mol%), and L3 (10 mol%) in toluene (1.0 mL) at r.t. for 4 h under an argon atmosphere. isolated yield. ee values were determined by chiral HPLC analysis. dr values were determined by crude 1H NMR analysis. | |
{kind=link}

|
Download:
|
| Scheme 3. Substrate scope for the [4 + 2] cycloaddition of enones 1 and 2b. | |
{kind=link}
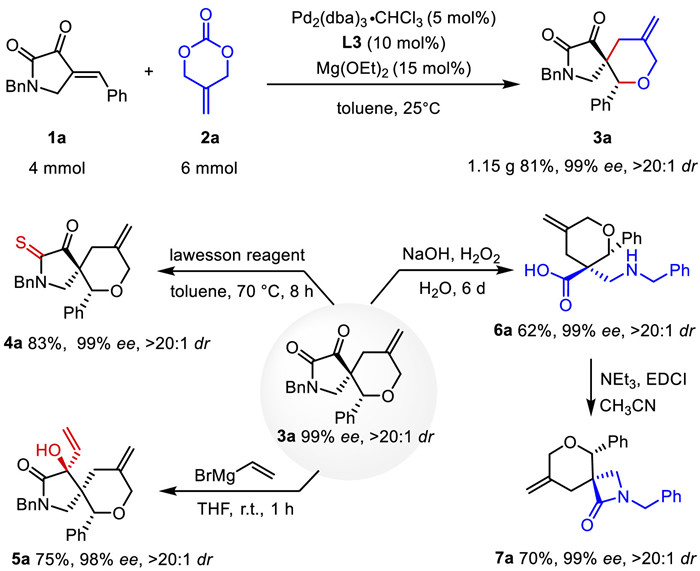
To demonstrate the synthetic applicability of the current [4 + 2] annulation, we performed a gram-scale reaction of 1a and 2a, yielding 1.15 g of 3a under optimized conditions. The potential utility of this method was further illustrated through the chemoselective transformation of individual functional groups. Treating 3a with Lawesson's reagent thionated the amide carbonyl group, generating 4a in 83% yield. Furthermore, vinylmagnesium bromide reacted at the other carbonyl site to produce 5a with three consecutive chiral carbon atoms and excellent diastereoselectivity. We also emphasized the transformation of 3a to 6a using H2O2. This regioselective Baeyer-Villiger oxidation facilitated the synthesis of a β2,2-amino acid bearing a quaternary stereocenter, which was otherwise difficult to access. The β2,2-amino acid 6a was then cyclized with EDCI to provide β-lactams 7a in 70% yield (Scheme 4) [23].

|
Download:
|
| Scheme 4. Scale-up reaction and further transformations of 3a. | |
{kind=link}
To gain some insight into the reaction pathway, several control experiments were conducted. First, we provided direct experimental proofs that the annulation reaction preferred to follow the stepwise pathway rather than the concerted pathway (Scheme S1 in Supporting information). Then, the formation of the [Pd-dba-L3] complex was detected by HRMS analysis. Additionally, we investigated the effect of the Pd/ligand ratio. Applying the method of continuous variation revealed a 1:1 binding interaction of Pd2(dba)3·CHCl3 and L3 in the presence of the standard reaction conditions (Fig. S2 in Supporting information) [24]. Meanwhile, the competitive reactions of alkene 1i and 1j with 1a were performed respectively under the standard reaction conditions, and the products 3i and 3j were obtained in 8% and 61% yields, respectively (3a/3i = 8.4:1, 3a/3j = 1.3:3, Scheme 5A). The substrate 1j bearing an electron-withdrawing group tended to be relatively more reactivity than substrate 1i possessing an electron-donating group. These results indicated that the cleavage of the carbon–carbon double bond, rather than the nucleophilicity of the corresponding carbocation, affected the reaction rates. Finally, a deuterium labeling experiment was conducted to elucidate further the mechanistic details of the transformation (Scheme 5B). The results revealed that intramolecular cyclization was a fast step, which was consistent with the conclusion of intermolecular competition experiments.

|
Download:
|
| Scheme 5. Experiments for mechanistic insights. | |
{kind=link}
Based on our experimental results and previous literature [11,25-29], a plausible catalytic cycle is proposed in Scheme 5C. 1,4-zwitterionic species Ⅰ is initially generated through decarboxylative oxidative insertion of palladium(0) to carbonates 2a. The oxygen anion of intermediate Ⅰ then undergoes Michael addition to enone 1a to generate the key intermediate Ⅱ. Due to the sterically crowded chiral ligand, a 1,4-conjugate addition of intermediate Ⅰ to the Re face of the enone 1a moiety occurs favorably. Allylic 1,3-strain may be viewed as a conformation-determining factor [30-33], conformation Ⅱ is strongly favored in the conformer equilibrium, while the high-energy conformer Ⅲ can usually be neglected. Subsequently, C—C coupling occurring at the terminal carbon of the π-allylpalladium in Ⅱ allows phenyl group to occupy equatorial orientation. After intramolecular cyclization, the desired product 3a is achieved with the release of the Lewis acid and palladium(0) catalyst for the next catalytic cycle.
In summary, the stereoselective construction of tetrahydropyran-fused spiro-pyrrolidine-2,3-diones in moderate to good yields with excellent enantioselectivities has been well established, providing contiguous quaternary and tertiary stereocenters. The chiral ligand (R)-DTBM-Segphos and allylic 1,3-strain were the key to constructing the chiral tetrahydropyran-fused spiro-pyrrolidine-2,3-diones. The synthetic utility of this method was demonstrated by synthetic transformations of the product.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentFinancial support from the National Key Research and Development Program of China (No. 2021YFF0600704) is gratefully acknowledged.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109157.
| [1] |
E. Vitaku, D.T. Smith, J.T. Njardarson, J. Med. Chem. 57 (2014) 10257-10274. DOI:10.1021/jm501100b |
| [2] |
C. Zhao, M. Sun, Y.L. Bennani, et al., J. Med. Chem. 51 (2008) 5423-5430. DOI:10.1021/jm8003625 |
| [3] |
A.K. Franz, P.D. Dreyfuss, S.L. Schreiber, J. Am. Chem. Soc. 129 (2007) 1020-1021. DOI:10.1021/ja067552n |
| [4] |
U. Hossain, A. Sharm, S. Ghosh, S. Bhattacharya, Eur. J. Med. Chem. 45 (2010) 3265-3273. DOI:10.1016/j.ejmech.2010.04.001 |
| [5] |
F. Thaler, L. Moretti, R. Amici, et al., Eur. J. Med. Chem. 108 (2016) 53-67. DOI:10.1016/j.ejmech.2015.11.010 |
| [6] |
O. Pàmies, J. Margalef, S. Cañellas, et al., Chem. Rev. 121 (2021) 4373-4505. DOI:10.1021/acs.chemrev.0c00736 |
| [7] |
B.M. Trost, G. Mata, Acc. Chem. Res. 53 (2020) 1293-1305. DOI:10.1021/acs.accounts.0c00152 |
| [8] |
P.W. Xu, J.S. Yu, C. Chen, et al., ACS Catal. 9 (2019) 1820-1882. DOI:10.1021/acscatal.8b03694 |
| [9] |
A.K. Franz, N.V. Hanhan, N.R. Ball-Jones, ACS Catal. 3 (2013) 540-553. DOI:10.1021/cs300801y |
| [10] |
B.M. Trost, P.J. Morris, Angew. Chem. Int. Ed. 50 (2011) 6167-6170. DOI:10.1002/anie.201101684 |
| [11] |
B. Mao, H. Liu, Z. Yan, et al., Angew. Chem. Int. Ed. 59 (2020) 11316-11320. DOI:10.1002/anie.202002765 |
| [12] |
Z.S. Liu, W.K. Li, T.R. Kang, L. He, Q.Z. Liu, Org. Lett. 17 (2015) 150-153. DOI:10.1021/ol503383x |
| [13] |
Y. Li, J. Jie, H. Li, H. Yang, H. Fu, Org. Lett. 23 (2021) 6499-6503. DOI:10.1021/acs.orglett.1c02306 |
| [14] |
Z. Chen, Z.C. Chen, W. Du, Y.C. Chen, Org. Lett. 23 (2021) 8559-8564. DOI:10.1021/acs.orglett.1c03279 |
| [15] |
C.V. Galliford, K.A. Scheidt, Angew. Chem. Int. Ed. 46 (2007) 8748-8758. DOI:10.1002/anie.200701342 |
| [16] |
E. Badiola, I. Olaizola, A.V. Zquez, et al., Chem. Eur. J. 23 (2017) 8185-8195. DOI:10.1002/chem.201700464 |
| [17] |
Y. Liu, Y. Zhang, Q.W. Huang, et al., Adv. Synth. Catal. 363 (2021) 2177-2182. DOI:10.1002/adsc.202100033 |
| [18] |
M. Fofana, Y. Dudognon, L. Bertrand, et al., Eur. J. Org. Chem. (2020) 3486-3490. DOI:10.1002/ejoc.202000460 |
| [19] |
Q.W. Huang, T. Qi, Y. Liu, et al., ACS Catal. 11 (2021) 10148-10158. DOI:10.1021/acscatal.1c01724 |
| [20] |
H. Wang, T. Zeng, W. Chang, L. Liu, J. Li, Org. Lett. 23 (2021) 3573-3577. DOI:10.1021/acs.orglett.1c00976 |
| [21] |
L. Liu, D. Guo, J. Wang, Org. Lett. 22 (2020) 7025-7029. DOI:10.1021/acs.orglett.0c02573 |
| [22] |
M. Wu, Z. Han, K. Li, et al., J. Am. Chem. Soc. 141 (2019) 16362-16373. DOI:10.1021/jacs.9b07418 |
| [23] |
A.Q. Cusumano, M.W. Boudreau, J.G. Pierce, J. Org. Chem. 82 (2017) 13714-13721. DOI:10.1021/acs.joc.7b02572 |
| [24] |
J.S. Renny, L.L. Tomasevich, E.H. Tallmadge, Angew. Chem. Int. Ed. 52 (2013) 11998-12013. DOI:10.1002/anie.201304157 |
| [25] |
R. Shintani, K. Moriya, T. Hayashi, Chem. Commun. 47 (2011) 3057-3059. DOI:10.1039/c0cc05308b |
| [26] |
P.H. Dou, S.P. Yuan, Y. Chen, et al., J. Org. Chem. 87 (2022) 6025-6037. DOI:10.1021/acs.joc.2c00276 |
| [27] |
L.C. Yang, Z.Y. Tan, Z.Q. Rong, et al., Angew. Chem. Int. Ed. 57 (2018) 7860-7864. DOI:10.1002/anie.201804160 |
| [28] |
M.L. Ke, Z.G. Liu, K. Zhang, S. Zuo, F.E. Chen, Green Synth. Catal. 2 (2021) 228-232. DOI:10.1016/j.gresc.2021.04.004 |
| [29] |
C. Zhao, I. Khan, Y.J. Zhang, Chem. Commun. 56 (2020) 12431-12434. DOI:10.1039/d0cc05640e |
| [30] |
R.W. Hoffmann, Chem. Rev. 89 (1989) 1841-1860. DOI:10.1021/cr00098a009 |
| [31] |
F. Johnson, Chem. Rev. 68 (1968) 375-413. DOI:10.1021/cr60254a001 |
| [32] |
S.K. Malhotra, F. Johnson, J. Am. Chem. Soc. 87 (1965) 5493-5495. DOI:10.1021/ja00951a047 |
| [33] |
R.W. Hoffmann, Angew. Chem. Int. Ed. 31 (1992) 1124-1134. |