 2024, Vol. 35
2024, Vol. 35 


Metal-nitrenoids are considered as important intermediates for the synthesis of nitrogen-containing compounds due to their unique reactivity [1–4]. Among different types of reactions developed, novel amination reactions via insertion of nitrene into C−H bond have been widely reported. Besides, the metal-nitrenoid intermediates undergo addition reaction when a proper nucleophile is contained, and interesting aziridination, olefin difunctionalization and sulfimidation reactions were disclosed. These methods provided new ways for the efficient construction of N−C and N−X (X = N, S) bonds under relatively mild conditions [5–7]. Hence, transition metal-catalyzed nitrene transfer reactions have received considerable attention.
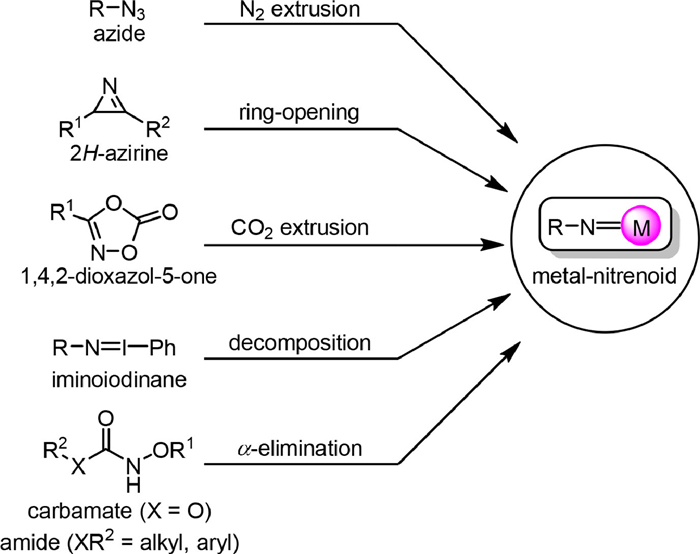
The metal-nitrenoid species could be generated from different precursors (Scheme 1). Traditionally, the azide derivatives were used as the nitrene precursors as they undergo nitrogen extrusion easily under photo-irradiation, heat, or transition metal promotion [8–11]. The 2H-azirines are highly strained molecules containing an unsaturated three-membered ring, and nitrene intermediates could be formed by ring-opening isomerization reactions under photo irradiation or transition metal catalysis [12–16]. Transition metal-catalyzed novel transformations of 1,4,2-dioxazol-5-ones were widely pursued [17–20], and the involvement of metal-nitrenoid was proposed as a result of the metal-induced decarboxylation. Primary amines and amides are inert for direct nitrene transfer reactions, however, iminoiodinane could be formed as an active species by oxidation with a hypervalent iodide reagent [21,22], which undergoes decomposition easily to form a metal-nitrenoid intermediate under transition metal catalysis. The α-elimination of N-arylsulfonoxy carbamates for generation of oxycarbonylnitrene was first reported in 1960s. In the past two decades, numerous applications of N-oxygen-substituted carbamates for C−H amination, alkene functionalization, and others were disclosed via transition metal-catalyzed nitrene transfer. More recently, the effective generation and trapping of the nitrene species from metal-triggered α-elimination of N-acyloxylamides was disclosed [23–33].

|
Download:
|
| Scheme 1. Versatile nitrene precursors. | |
{kind=link}
The developments in nitrene transfer reactions with azide derivatives [34–36], iminoiodinane [37], 2H-azirine [38], and 1,4,2-dioxazol-5-one have been previously reviewed by different groups [39]. To showcase the unique reactivity of carbamates as precursors for the synthesis of complex nitrogen-containing molecules, herein, the recent advances in transition metal-catalyzed nitrene transfer with carbamate derivatives were overviewed based on different types of reaction, in which the metal-nitrenoid intermediates could be involved by decomposition of the in-situ generated iminoiodinanes from primary carbamates or by α-elimination of secondary carbamates containing an N-oxygen substituent. It should be noted that the N-alkoxycarbonyl azides are also carbamate derivatives, which may undergo nitrene transfer reactions as normal azides by nitrogen extrusion [40–44]. Such reactions were not included in this review.
2. Catalytic C(sp3)−H aminationCatalytic C(sp3)−H amination involving nitrene intermediates has emerged as an important alternative to classical methods for C(sp3)−N bond formation [45–47]. In this respect, the intramolecular C(sp3)−H amination of carbamates has been developed for the effective construction of oxazolidinones and other heterocycles.
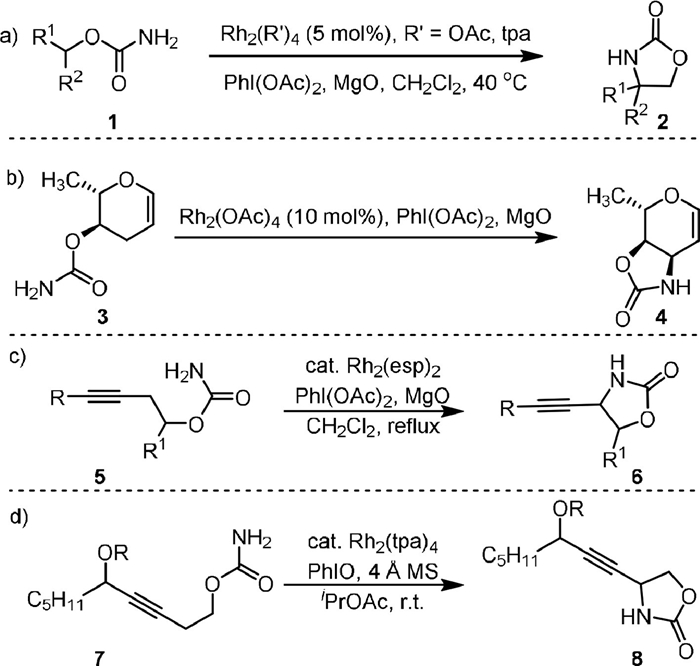
In 2001, the Du Bois’ group reported the first intramolecular oxidative cyclization of primary carbamates (1) to oxazolidinones (2) using Rh(Ⅱ) carboxylate complexes as the catalyst and PhI(OAc)2 as the oxidant (Scheme 2a) [48]. The addition of MgO as an AcOH scavenger is important for high yield transformation, as initial condition screening indicated that the generated by-product AcOH might reduce the activity of the catalyst. In some cases, the complex with triphenylacetate (tpa) anion showed enhanced performance due to its greater resistance towards oxidation under the reaction conditions. When the enantioenriched primary carbamate was used as the substrate, the resulting oxazolidinone maintained the configuration without racemization, indicating that the formation of the C−N bond was a stereospecific process, which involves direct nitrene or nitrenoid insertion rather than radical species. The synthetic utility of this stereospecific Rh-nitrene C−H insertion was demonstrated by its application for the total synthesis of (-)-tetrodotoxin in a later report [49].

|
Download:
|
| Scheme 2. Rhodium-catalyzed intramolecular C(sp3)−H amination of carbamates. | |
{kind=link}
A DFT study conducted by the Che group [50] in 2007 provided more detailed information about the mechanism of the above reaction. It was showed that the concerted singlet pathway is more favorable than the triplet pathway according to the calculated free energy of activation, thus leading to the retention of the configuration of the carbon atom in the product.
In 2005, Parker [51] applied the Du Bois’ oxidative cyclization for the synthesis of carbamate-protected glycals of naturally occurring 3,4-cis-3-amino-2,3,6-trideoxyhexoses (4) from noncarbohydrate starting materials 3 (Scheme 2b). The reaction was found to be highly chemoselective, as the insertion of a rhodium nitrene in an allylic C−H bond is more favorable than the insertion in a C−H bond that is α to an oxygen substituent.
In 2012, Schomaker [52] reported the Rh-catalyzed synthesis of propargyl amines (6) from homopropargyl carbamates (5, Scheme 2c). The target product could be obtained under the conditions of Rh2(esp)2 as catalyst, PhI(OAc)2 as oxidant in CH2Cl2 under reflux. The protected propargyl alcohol carbamates (8) could be generated from propargyl alcohol substrates (7) when Rh2tpa4 was used as the catalyst and PhIO was used as the oxidant (Scheme 2d). After deprotection, 8 could be converted to allenic amines under the Myers’ condition [53].
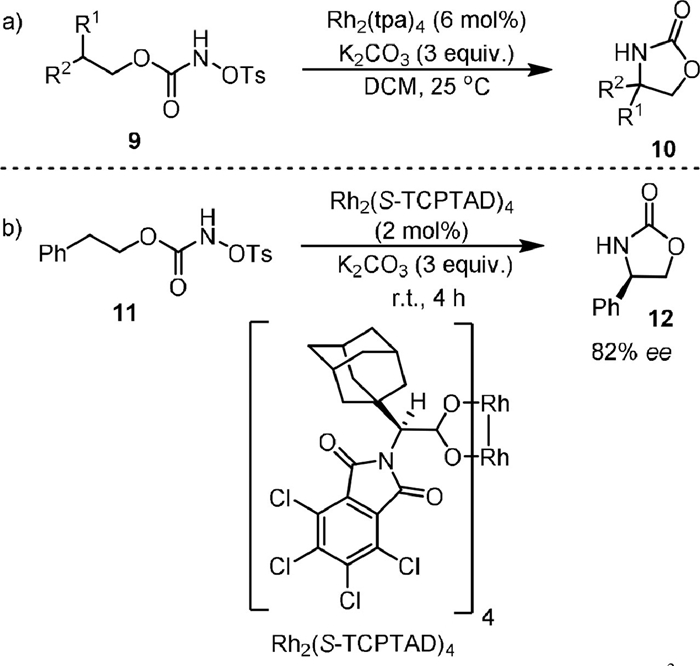
The using of external oxidant could be avoided when N-tosyloxycarbamates are used as the substrates. In this respect, in 2005 the Lebel group [54] reported the intramolecular cyclization of N-tosyloxycarbamates (9) with 6 mol% of Rh2(tpa)4 as the catalyst and 3 equiv. of K2CO3 as the base, generating the oxazolidinones (10) in DCM (Scheme 3a). By carrying out the reaction with chiral substrate, they found that the formation of C−N bond was stereospecific. The amination method was found to be applicable for different substrates containing ethereal, benzylic, tertiary, secondary, and even primary C−H bonds [55]. The stereospecific C−N bond formation was also observed in the intramolecular oxidative cyclization of primary carbamates as reported by Yakura [56]. The first intramolecular enantioselective C−H aminations was reported in 2006 by Davies and coworkers (Scheme 3b) [57]. They developed the Rh2(S-TCPTAD)4 as a new chiral Rh catalyst for converting N-tosyloxycarbamates (11) to oxazolidinones (12) with good enantioselectivity (82% ee). Notably, they found the Rh2(S-TCPTAD)4 could also be used for the intermolecular C−H aminations of indane derivations with NsNH2 with high enantioselectivity (up to 94% ee).

|
Download:
|
| Scheme 3. Rhodium-catalyzed intramolecular C(sp3)−H amination of N-tosyloxycarbamates. | |
{kind=link}
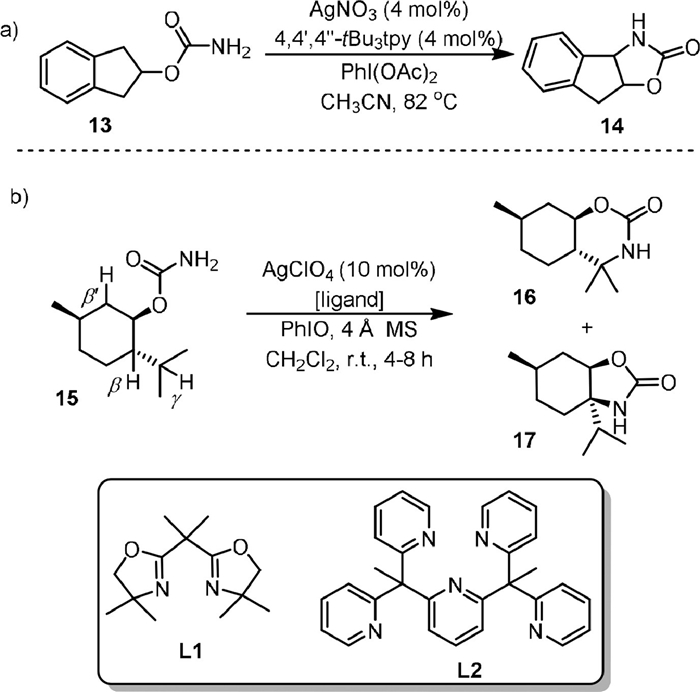
Except for rhodium catalyst, examples with complexes of other metals were known for intramolecular C(sp3)−H amination of carbamates. In 2004, He [58] used AgNO3-4,4’,4’’-tBu3tpy as the catalyst and PhI(OAc)2 as the oxidant for converting the carbamates (13) to the five-membered ring products (14) in good to excellent yields (Scheme 4a). It was found that only the 4,4’,4’’-tBu3tpy ligand showed good results, and the catalysts with other types of ligands were either inactive or resulted in low yields. Controlled experiments showed that the reaction was stereospecific and the insertion of nitrene into C−H bonds was most likely involved.

|
Download:
|
| Scheme 4. Silver-catalyzed intramolecular C(sp3)−H amination. | |
{kind=link}
In 2019, Schomaker [59] reported a regioselectivity-controlled intramolecular C−H bonds amination of carbamates by changing the ligand on the silver catalyst. As exemplified by the reaction of 15 in Scheme 4b, when the less sterically hindered bis(oxazoline) ligand L1 was used, the AgClO4-L1 complex favored γ-C−H amination of the menthol (15) and produced the six-membered oxazinanone ring product 16 in 84% yield, while the five-membered oxazolidinone ring product 17 from β-C−H amination was obtained as a minor product (15% yield). When the more sterically hindered 2, 6-bis[1, 1-bis(2-pyridyl)ethyl]-pyridine ligand L2 was used, the five-membered oxazolidinone ring product 17 was reported as the sole product from the β-C−H amination. These two catalysts resulted in the same regioselectivity for the amination of a wide range of carbamate substrates. The reactions utilizing these two catalysts could provide valuable 1,2- and 1,3-aminoalcohols at the gram-scale in excellent yields and could be used for late-stage functionalization of drug precursors and natural products.
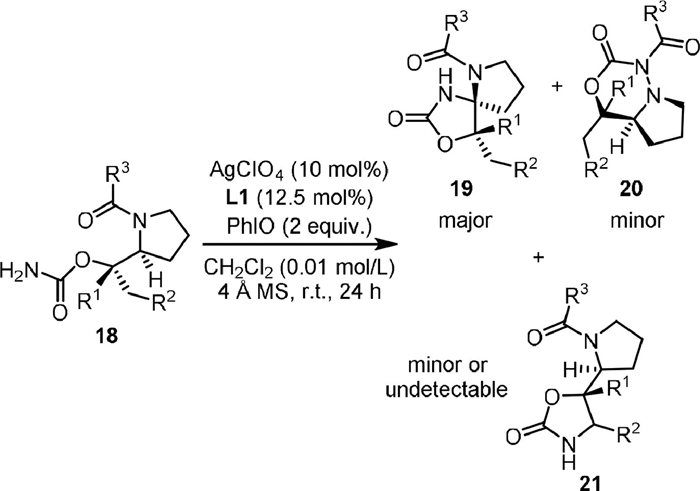
In 2020, Harada and Nemoto [60] reported a site- and chemo-selective C−H functionalization to synthesize densely functionalized spiroaminals (19, Scheme 5), in which the AgClO4-L1 complex and PhIO served as the catalyst and the oxidant, respectively. Challenging substrates with multiple reaction sites could be converted to spiroaminals with high site-selectivity and chemoselectivity. Both AgBF4-L1 and AgOTf-L1 showed similar catalytic activity, but the desired product could not be obtained in the absence of silver catalyst or replacing the PhIO with PhI(OAc)2 or PhI(OPiv)2. Theoretical and experimental mechanistic studies suggested that the chemoselective C−H bond amination for generation of stereodefined spiromolecules occurs through an asynchronous concerted process involving triplet spin-correlated radical pairs. This work showed the reaction with silver-based catalytic system is different from the rhodium catalysis, which generally gave amide C−N bond insertion products.

|
Download:
|
| Scheme 5. Silver-catalyzed synthesis of spiroaminals via C−H functionalization. | |
{kind=link}
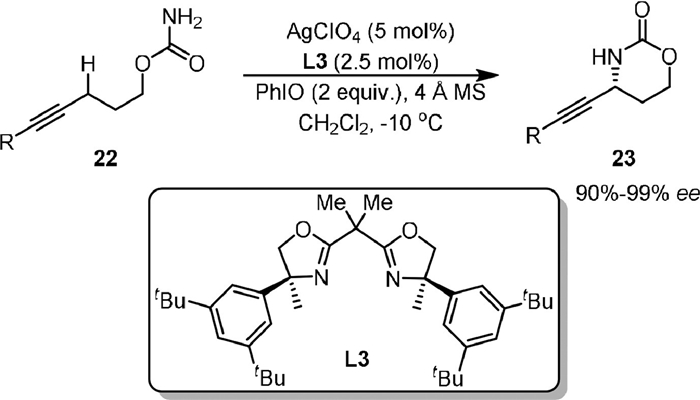
In 2020, Schomaker developed a silver-catalyzed enantioselective propargylic C−H amination for transforming carbamate esters bearing two prochiral γ-propargylic C−H bonds (22) to enantioenriched γ-alkynyl γ-aminoalcohols (23, Scheme 6). In this protocol a new bis(oxazoline) (BOX) ligand (L3) was designed via a rapid structure−activity relationship (SAR) analysis [61]. The hydrogen-atom transfer (HAT) from the putative silver-nitrene radical anion was proposed as the enantiodetermining step of the reaction, which was understood by density functional theory calculations.

|
Download:
|
| Scheme 6. Synthesis of propargylic carbamates via the C−H amination of alkynes. | |
{kind=link}
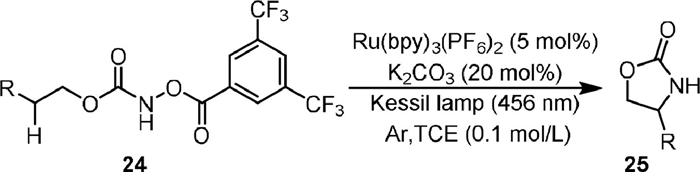
Ruthenium complexes have also been reported as catalysts for the selective intramolecular C(sp3)−H amination of carbamates. In 2020, Chang [62] designed a photosensitization strategy to generate triplet nitrenes from hydroxamates bearing an N-[3, 5-bis(trifluoromethyl)benzoyl] moiety, which was applied for the intramolecular C−H amidation reactions (Scheme 7). In the presence of Ru(bpy)3(PF6)2 as a photosensitizer and K2CO3 as a base under visible light irradiation, N-benzoyloxycarbamate (24) could be transformed into oxazolidinone (25) with high functional group compatibility. Mechanistic investigations indicated the reaction proceeded via a triplet energy transfer pathway.

|
Download:
|
| Scheme 7. Ru-catalyzed intramolecular C(sp3)−H amination of hydroxamates. | |
{kind=link}
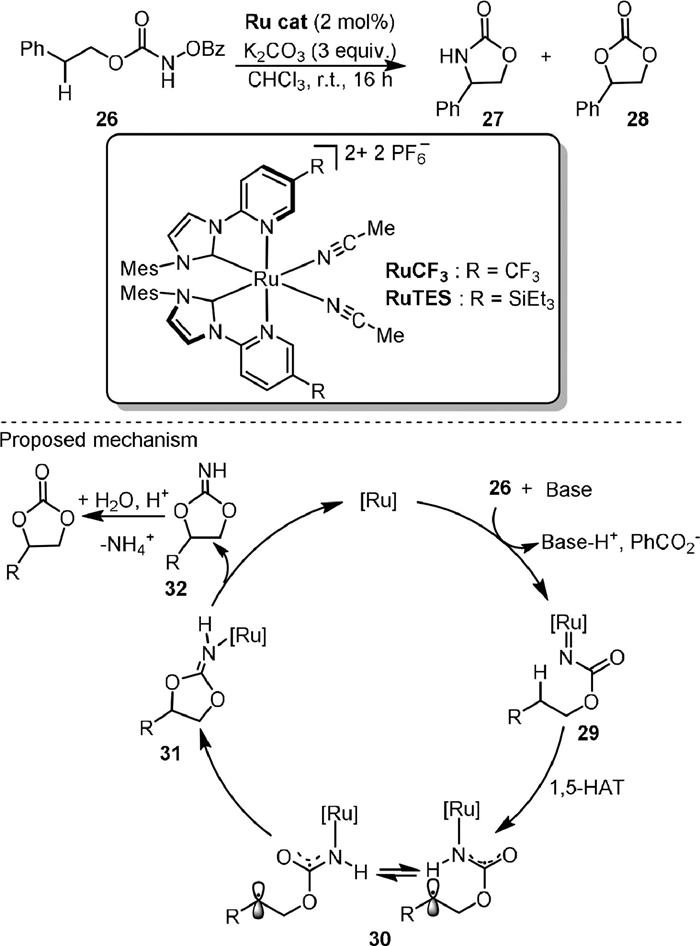
In the same year, Houk and Meggers [63] used the ruthenium complexes containing two bidentate N-(2-pyridyl)-substituted N-Heterocyclic carbenes and two acetonitrile ligands as the catalysts for controllable intramolecular C−H bond amination and oxygenation (Scheme 8), as exemplified by the reaction of N-benzoyloxy carbamates (26) to oxazolidinone (27) and cyclic carbonate (28), respectively. The chemoselectivity was controlled by R substituent of the catalyst. When RuCF was used, the amination product 27 could be obtained in 87% yield. When RuTES was used, the oxygenation product 28 was obtained as the major product. An enantioselective fashion of reaction was also developed. Based on the experimental and computational results, it was proposed that the triplet state ruthenium nitrenoid intermediate (29) first underwent 1,5-hydrogen atom transfer to form the diradical species 30. If the lifetime of 30 was long enough, it could achieve conformational reorganization prior to the radical-radical recombination for C-O bond formation (31). From the protonated intermediate iminocarbonate 32, the oxygenation product could be formed by hydrolysis.

|
Download:
|
| Scheme 8. Rutheuium-catalyzed intramolecular C(sp3)−H amination and oxygenation. | |
{kind=link}
Intermolecular C(sp3)−H amination using carbamate as the nitrene precursor has been much less explored. In 2007, Lebel [64] used Rh(tpa)4 as a catalyst to realize the first intermolecular C−H amination (Scheme 9). The reaction used 2,2,2-trichloroethyl-N-tosyloxycarbamate (34) as the nitrene source, which reacted with cyclic aliphatic alkanes or aromatic alkanes for the synthesis of a variety of Troc-protected amines. It is notable that the nitrene insertion could proceed in primary C−H bonds and an enantioselective version of transformation was developed when chiral rhodium catalysts were used.

|
Download:
|
| Scheme 9. Rhodium-catalyzed intermolecular C(sp3)−H amination with N-tosyloxycarbamates. | |
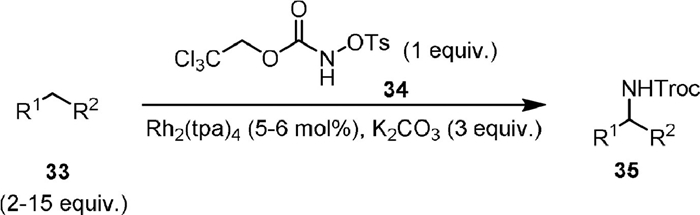{kind=link}
In 2011, the same group [65] reported a stereoselective intermolecular allylic C−H amination of E-β-ethylstyrenes by using R-1-phenyl-2,2,2-trichloroethyl-N-tosyloxycarbamate (36) as the chiral nitrene precursor and Rh2[(S)-Br-nttl]4 as the chiral catalyst (Scheme 10). The absolute stereochemistry of the product was determined by X-ray crystallographic study. Facile remove of the Ph-Troc protecting group was carried out to recover the free amine in high yields without degradation.

|
Download:
|
| Scheme 10. Rhodium-catalyzed stereoselective intermolecular C(sp3)−H amination of alkenes. | |
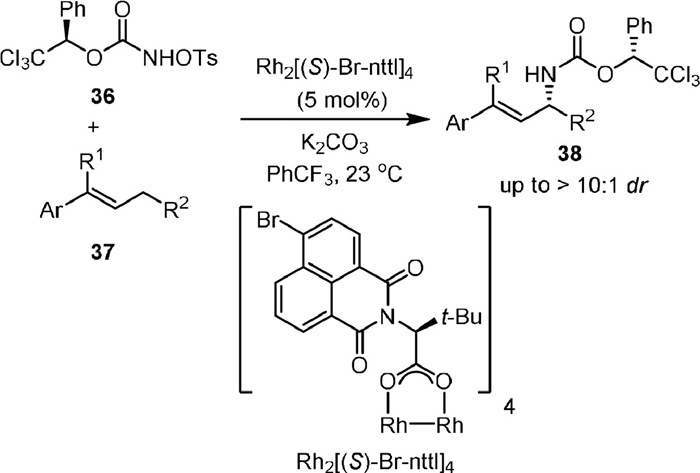{kind=link}
Very recently, the Chang group reported a high-throughput experimentation for developing a series of tailored CpXRh(LX)Cl catalyst systems for the outer-sphere C−H amidations. By using N-tosyloxycarbamates as the nitrenoid precursor, the optimal CpXRh(Ⅲ)(LX) catalysts were quickly identified for intra- and intermolecular C−H amidations and also for an enantioselective transformation [66].
3. Directed C(sp2)−H aminationThe intermolecular nitrene transfer reaction for C−H amination of aromatic compounds is a powerful means to access synthetically important aromatic amines. With the assistance of a proper directing group, selective C−H amination of different aromatic compounds with carbamates has been achieved [3,47,67].
In 2010, Yu [68] developed a novel method for the synthesis of 2-aminoanilines (41) by the Pd(Ⅱ)-catalyzed direct intermolecular ortho-C−H amination of anilides (41) with N-nosyloxycarbamates (40, Scheme 11a). Amide groups were used as the directing group and only monoamidation products were obtained exclusively in all cases. It was found that the N-pentafluorobenzoate (OPFB) and N-pivalate (OPiv) carbamates were ineffective reagents for the reaction and N-tosyloxycarbamate showed low reactivity. The reaction could be carried out under relatively mild conditions with high functional group tolerance and regioselectivity. Kinetic isotope effect (KIE) experiments showed that C−H activation was a rate-determining step (KH/KD = 3.7).

|
Download:
|
| Scheme 11. Palladium-catalyzed intermolecular C(sp2)−H amination. | |
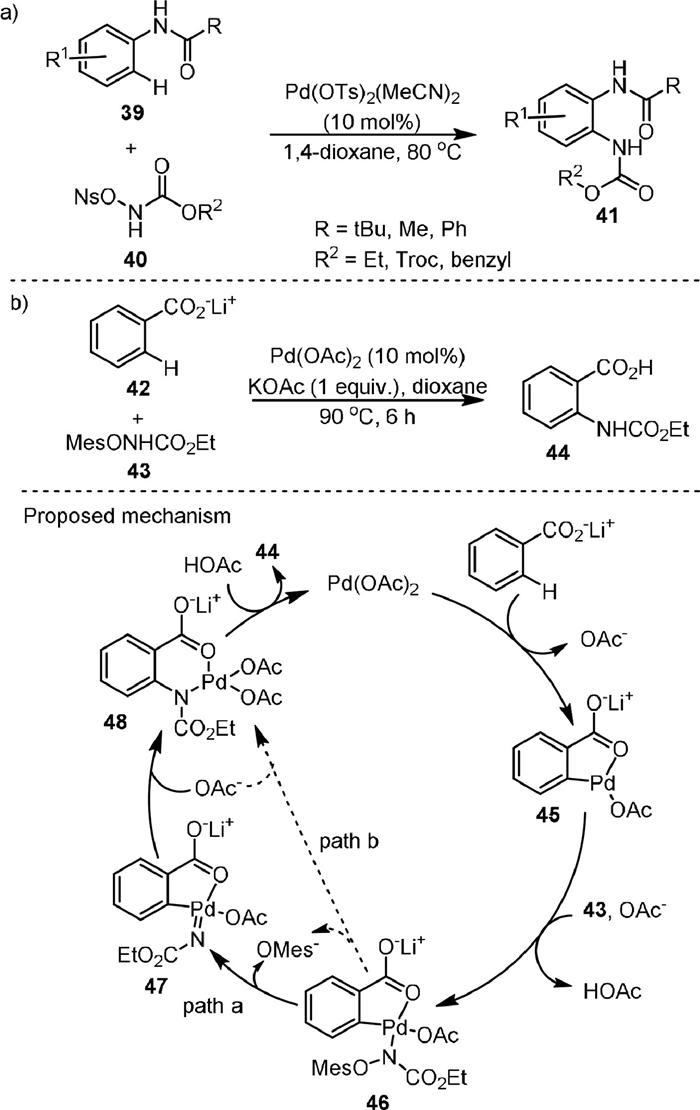{kind=link}
In 2012, the same group [69] reported a convenient route to anthranilic acid (44) via carboxylate-assisted ortho-C−H amination of benzoates (42) with mesitylsulfonate (43) in the presence of Pd(OAc)2 as the catalyst and KOAc as the additive (Scheme 11b). The counter ion of 42 was found to be critical for the reaction, and Na+, K+ and N(n-Bu4)+ produced lower product yields than Li+. The proposed mechanism suggested that the cyclopalladation of 42 with the catalyst produced intermediate 45, and the KH/KD value from KIE study was 2.6, indicating that the C−H bond cleavage could be probably involved as a rate-determining step. Next, the N-mesitylsulfonyloxycarbamate anion reacted with 45 to form intermediate 46. From 46, the C−N bond formation for the generation of intermediate 48 could possibly occur via a stepwise process involving the elimination of OMes anion to form 47 and the migratory insertion of Pd-nitrene species to Pd-C bond (path a), or a concerted aryl migration with the departure of the sulfonate (path b). Finally, the product was formed by protonation of 48. The proposed five-membered cyclopalladation intermediate was previously isolated in a related study by Giri and Yu [70].
In 2013, Glorius [71] reported the synthesis of N-Boc protected arylamines by Rh(Ⅲ) catalysis (51, Scheme 12a). It was found that both the pyridine and O-methyl hydroxamic acids were effective directing groups (49), and the electron-deficient 2, 4, 6-trichlorobenzoyl group was the optimal leaving group of aroyloxycarbamates (50). This reaction has the advantage of mild conditions and broad functional group tolerance.

|
Download:
|
| Scheme 12. Rhodium-catalyzed C(sp2)−H amination. | |
{kind=link}
In the same year, Zhou and Yang [72] reported the Rh-catalyzed amination of arene (52) with readily available N-hydroxycarbamates (53, Scheme 12b). Pyridine, pyrimidine, pyrazole, and N-OMe oxime could be employed as directing groups in this process, providing a variety of N-carbamate protected arylamines. The protecting group could be Cbz, Moz, Ac, Boc or even Fmoc.
In 2014, Chang [73] reported the Ir-catalyzed C−H amination of arenes (55) with carbamates (56) to afford N-substituted anilines (57, Scheme 13) in high efficiency with excellent functional group tolerance. Heterocyclic compounds including pyridine, pyrimidine, pyrazole, oxazoline could be used as the directing groups in this approach. The reaction could proceed smoothly at room temperature without the addition of external oxidants or bases. A small value of KH/KD (1.19) indicated that C−H activation was not a rate-limiting step. It was worth noting that in addition to aroyloxy- and acyloxycarbamates, other hydroxylamines containing Bz, Ts or Ac groups could also undergo the reaction effeciently. Soon after, the same group [74] completed a similar reaction using CoCp*(CO)I2 as the catalyst, in which only the O-acetoxycarbamates (58) could be used as a convenient amidating reagent. The reaction formed the acetic acid as the by-product which could be easily removed. It is worth noting that the purines could also be used as the directing groups of this reaction, and the formed 6-arylpurines bearing sensitive functional groups were biologically interesting.

|
Download:
|
| Scheme 13. Iridium and cobalt-catalyzed C(sp2)−H amination. | |
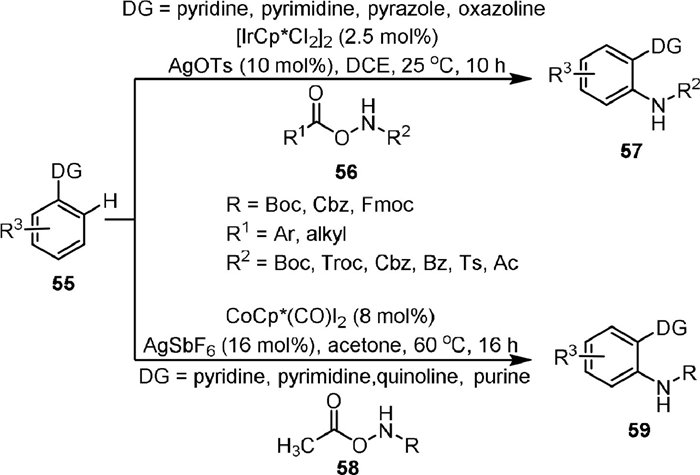{kind=link}
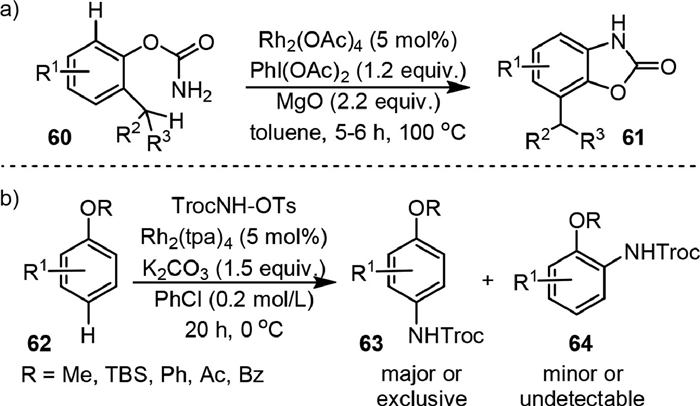
Intramolecular C(sp2)−H amination could be achieved with arylcarbamates. In 2016, Singh group [75] reported the Rh(Ⅱ)-catalyzed C−H insertion of O-phenyl carbamate (60) to yield benzoxazolones (61) in the presence of PhI(OAc)2 and MgO (Scheme 14a). It was found that the reaction was highly chemoselective for C(sp2)−H bond amination rather than more labile o-C(sp3)−H. Inverse secondary kinetic isotope effect was observed in mechanistic investigation (PH/PD = 0.42 ± 0.03), indicating this aryl C−H amidation transformation proceeded via the aromatic electrophilic substitution mechanism.

|
Download:
|
| Scheme 14. Rhodium-catalyzed aromatic C(sp2)−H amination. | |
{kind=link}
In 2018, Ueda and Kawabata [76] used O-tosyl-N-trichloroethoxy-carbonylhydroxylamine (TrocNH-OTs) as the nitrene precursor and Rh2(tpa)2 as the catalyst to realize the C−H amination of alkoxyarenes (62) in a chemo- and regioselective manner (Scheme 14b). The para-C(sp2)−H amination was more favorable than that of the benzylic C(sp3)−H bonds and/or α-C(sp3)−H bonds of the ethereal oxygen. Mechanistic insights based on KIE and radical clock experiment showed that C−H bond cleavage was not the rate-determining step (KH/KD = 1.16), and radical intermediates were not involved in the reaction path.
4. AziridinationAziridines are the smallest nitrogen-containing heterocycles, which are important synthetic targets and building blocks in synthesis of complex molecules [77,78]. Such compounds could be conveniently synthesized by the addition of metal-nitrenoid to alkenes.
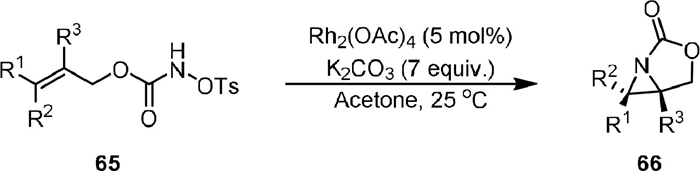
In the study of nitrene species generated from N-tosyloxycarbamates under Rh(Ⅱ) catalysis, an intramolecular aziridination reaction was observed by Lebel in 2005 (Scheme 15) [55]. The allylic N-tosyloxycarbamates (65) could be converted to aziridines (66) using 5 mol% of Rh2(OAc)4 with 3 equiv. of K2CO3 in acetone solvent. Both disubstituted and trisubstituted alkenes worked well under the conditions, forming the corresponding aziridines. The aziridination reaction is stereospecific, as only one diastereomer was observed.

|
Download:
|
| Scheme 15. Rhodium-catalyzed aziridination reaction of N-tosyloxycarbamates. | |
{kind=link}
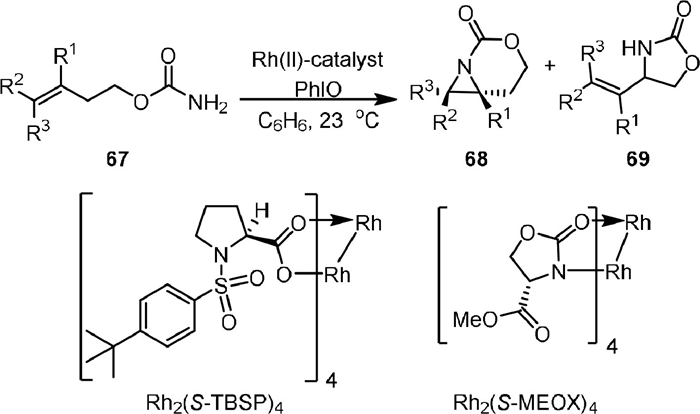
In 2006, Hayes [79] reported the Rh-catalyzed aziridination and 1,5-C−H insertion reactions of homoallyl carbamates 67 in the presence of PhIO (Scheme 16). It was found that the aziridination was favored over the competing C−H insertion when Rh2(S-TBSP)4 was used as catalyst, affording 68 as the major product with a stereospecific manner. However, when Rh2(S-MEOX)4 was used, the 1,5-C−H insertion product 69 was obtained. Other catalysts, such as Rh2(OAc)4 and Rh2(Oct)4, afforded both products in poor selectivity, suggesting the ligand played an important role in controlling the chemoselectivity of the reaction.

|
Download:
|
| Scheme 16. Rhodium-catalyzed aziridination and C−H amination of homoallyl carbamates. | |
{kind=link}
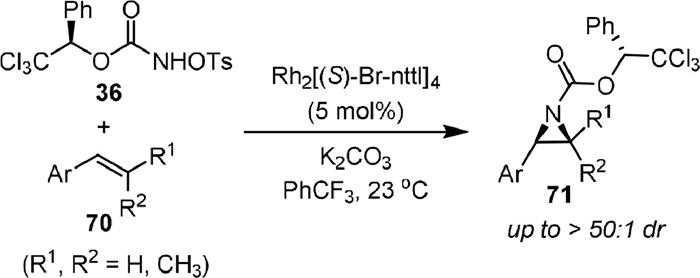
The first intermolecular stereoselective aziridination of alkenes (70) with R-1-phenyl-2,2,2-trichloroethyl-N-tosyloxycarbamate 36 was reported in 2011 by Lebel (Scheme 17) [65]. The rhodium complex Rh2[(S)-Br-nttl]4 was used as a chiral catalyst, and the absolute stereochemistry of the product 71 was determined by X-ray crystallographic study. It was shown that the formed chiral carbamate-protected aziridines was easily deprotected to form free aziridine and chiral alcohol under basic conditions.

|
Download:
|
| Scheme 17. Rhodium-catalyzed stereoselective intermolecular aziridination alkenes. | |
{kind=link}
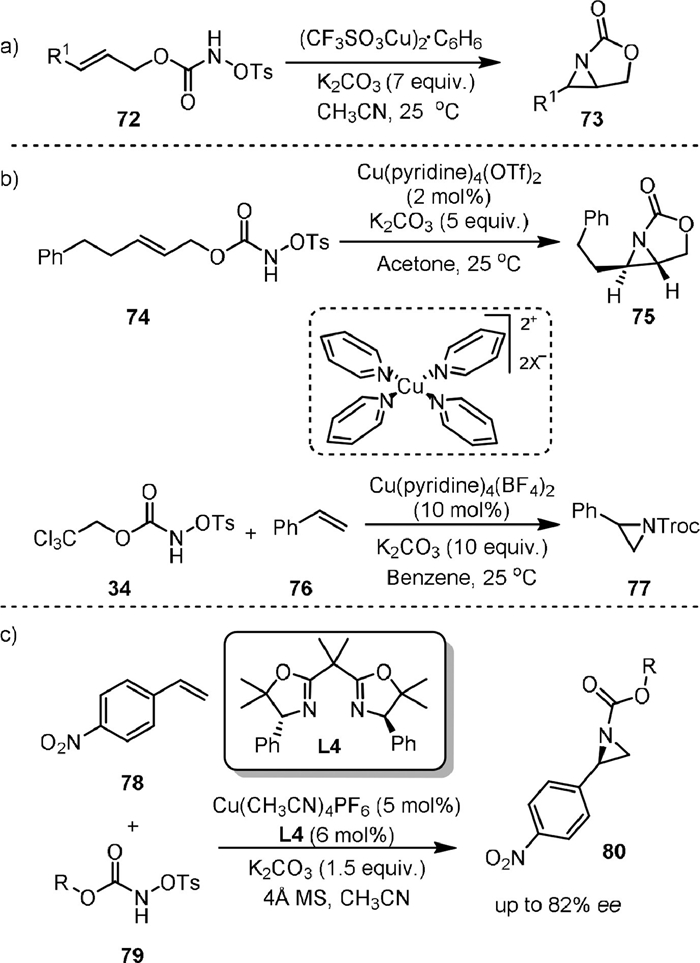
Copper complexes are much less expensive than Rh(Ⅱ) dimer and could also be used as capable catalysts for the intra- and intermolecular aziridination of alkenes with carbamates. In 2007, Fleming [80] reported the Cu-catalyzed aziridination of allylic N-sulfonyloxy carbamates 77 (Scheme 18a), which was applied for the synthesis of a 1,2-diamino-3-hydroxycyclohexane. An excess amount of base was found to be necessary. The bicyclic product 73 could undergo nucleophilic addition reaction with nucleophiles including RSH, R2NH, N3− and ROH.

|
Download:
|
| Scheme 18. Copper-catalyzed aziridination of alkenes. | |
{kind=link}
In the same year, Lebel [81] achieved the intramolecular aziridination of allylic N-tosyloxycarbamates and the intermolecular aziridination of styrenes with trichloroethyl N-tosyloxycarbamates using pyridine copper complexes as active catalysts (Scheme 18b). The intramolecular aziridination of 74 led to product 75 when using Cu(pyridine)4(OTf)2 as a catalyst in acetone. A stepwise mechanism involving presumably a triplet metal nitrene intermediate was proposed, as a mixture of cis- and trans-aziridines was obtained from the aziridination of Z-disubstituted allylic N-tosyloxycarbamates [82,83]. The intermolecular aziridination between trichloroethyl N-tosyloxycarbamate (34) and styrene was realized by using Cu(pyridine)4(BF4)2 as the catalyst in benzene, yielding aziridine 77 efficiently. Good chemoselectivity was obtained, as the aziridination of the benzene ring and the nitrene C−H insertion at the benzylic methyl group were not observed. A later work by the same group reported the enantioselective aziridination of 4-nitrostyrene (78) with N-tosyloxycarbamates (79) by using Cu(CH3CN)4PF6 in-combination with the gem-dimethyl-phenylglycine derived-bisoxazoline L4 (Scheme 18c) [84].
Interestingly, the aziridine could also be formed by intramolecular reaction of primary carbamates in the presence of PhI=O without metal catalyst according to the report by Padwa [82,83].
5. Bifunctionalization of alkenesThe aziridine is highly reactive and undergoes ring-opening reactions easily with nucleophiles under metal-free conditions, leading to products of formal alkene bifunctionalization in an anti-addition manner [85,86]. When metallonitrene species are formed from transition metal-catalyzed reactions of carbamates, diverse intra- and intermolecular alkene bifunctionalizations were developed in presence of different nucleophiles.
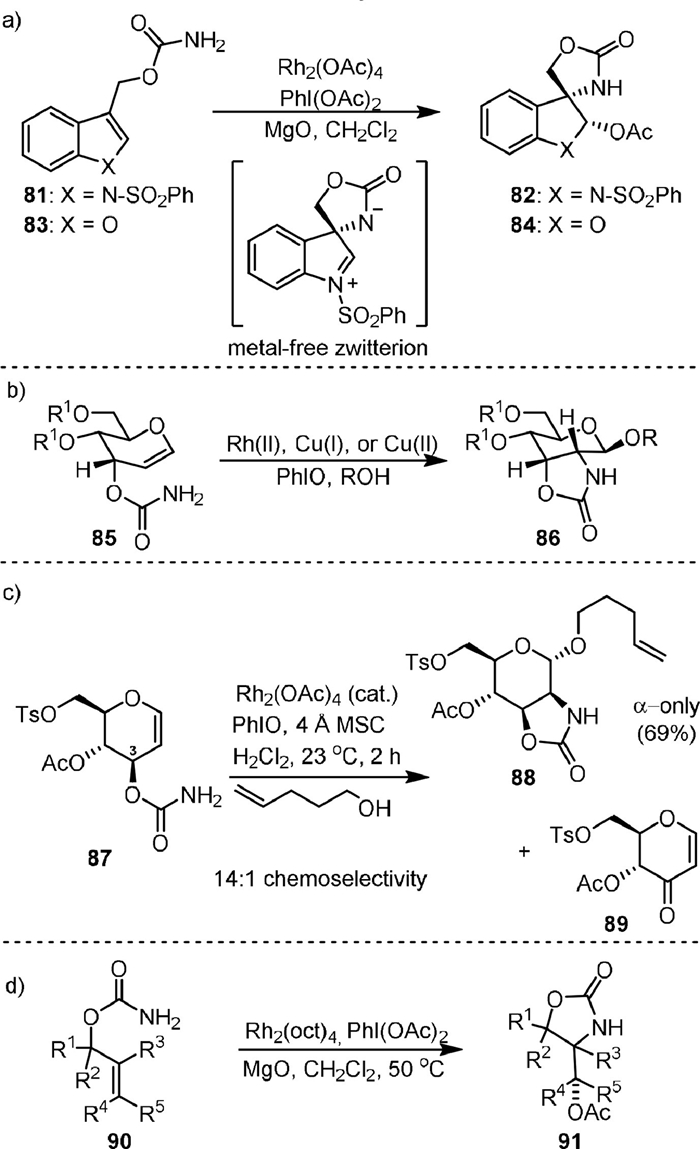
In 2002, Padwa [85] reported the rhodium(Ⅱ)-catalyzed reactions of allyl-substituted primary 3-indolyl carbamates 81 in the presence of PhI(OAc)2 (Scheme 19a), which formed compounds 82 by simultaneous spirocyclization of C3 and stereoselective acylation at C2. A metal-free zwitterionic intermediate by intramolecular addition of the nitrene species to the double bond was proposed, as the stereoselective formation of the product from syn-addition of the acetate group on the same side of the amide anion was observed. Later, they extended the reaction to 2-indolyl- and 3-benzofuranyl-substituted carbamate (83) to generate the corresponding spirocyclization product (84) with lower stereoselectivity [86].

|
Download:
|
| Scheme 19. Rhodium-catalyzed aminohydroxylation of allyl-substituted carbamates. | |
{kind=link}
Almost at the same time, Rojas [87] reported the metal-catalyzed amidoglycosylation of allylic allal 3-carbamates (85) in the presence of iodosobenzene. Upon the generation of a metal-complexed glycosyl aziridine intermediate, the 2-amino sugar (86) may be formed via sequential C2−N bond formation and β-selective glycosylation (Scheme 19b). The metal catalyst could be Rh2(OAc)4, Cu(MeCN)4PF6, or Cu(acac)2.
In 2009, the same group [88] reported the Rh-catalyzed amidoglycosylation of glucal 3-carbamates (87, Scheme 19c) to form 2N, 3O mannosamine oxazolidinones (88). High α-anomer selectivity could be achieved by acyclic 4O and 6O protecting groups with further improvement in less polar solvents. The byproducts (89) from C3−H oxidation could be inhibited by electron-withdrawing 4O and 6O protecting groups. Stereo- and chemoselectivity in this reaction reflect an interplay of stereoelectronic, conformational, and inductive effects. In 2014, Robertson [89] used the more organic soluble Rh2(oct)4 as the catalyst to transform a variety of allylic carbamates (90) into 4-acetoxymethyl-1,3-oxazolidin-2-one derivatives (91) with moderate to high levels of stereoselectivity (Scheme 19d).
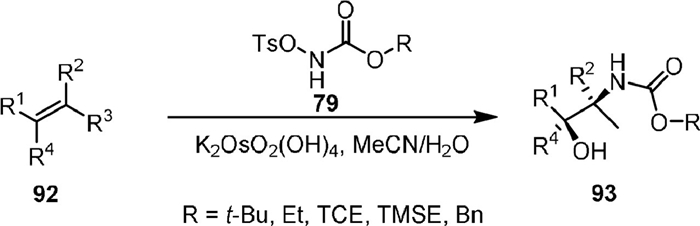
In 2012, McLeod [90] reported the osmium-catalyzed vicinal oxyamination of alkenes (92) with N-(tosyloxy)carbamates (79) to afford vicinal amino alcohol (93, Scheme 20). The reaction was applicable for a variety of mono-, di-, and tri-substituted alkenes. The loading of the osmium catalyst could be lowered to 0.1 mol% but a longer reaction time was required. Good regioselectivity was obtained for unsymmetrically substituted alkenes.

|
Download:
|
| Scheme 20. Osmium-catalyzed vicinal oxyamination of alkenes. | |
{kind=link}
Inexpensive iron complexes can also be used as catalysts for the bifunctionalization of alkenes with carbamates as the nitrogen sources, and interesting new reactions were disclosed by the Xu group [91–98].
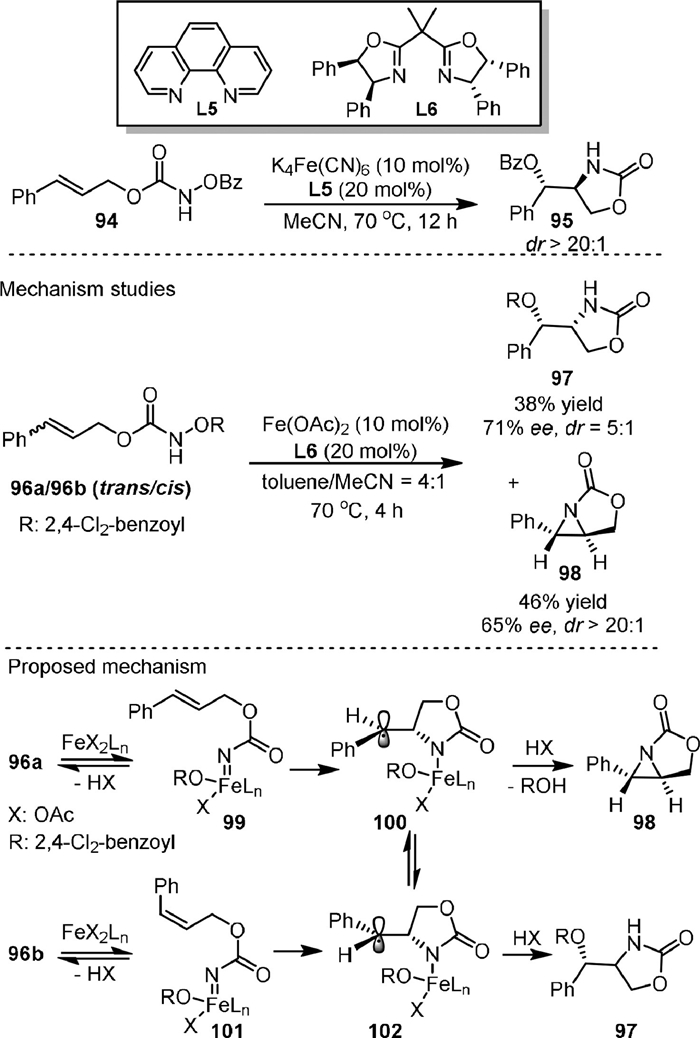
The first example of Fe-catalyzed intramolecular aminohydroxylation of olefins was reported in 2013 (Scheme 21) [91]. They reported the transformation of functionalized hydroxylamines (94) into synthetically useful amino alcohols (95) with high diastereoselectivity (dr up to > 20:1). The optimal catalyst was K4Fe(CN)6 in-combination with the 1, 10-phenanthroline ligand L5. Mechanistic studies showed that both 96a (trans) and 96b (cis) could be converted to the syn−hydroxyl oxazolidinone (97) and chiral aziridine (98) in the presence of a complex from Fe(OAc)2 and chiral bisoxazoline ligand L6. More importantly, the sense of enantioinduction was the same in both reactions with cis or trans substrates, which meant that the same intermediate was formed in both reactions.

|
Download:
|
| Scheme 21. Iron(Ⅱ)-catalyzed intramolecular aminohydroxylation of olefins. | |
{kind=link}
According to the proposed mechanism, firstly, 96a and 96b underwent N-O bond cleavage under the action of catalyst to generate iron nitrenoids 99 and 101, respectively. Next, a stepwise cycloamination process occurred via carbo-radical species 100 and 102, respectively, to generate 98 and 97. The formation of the same products from both substrates could be the results from the fast equilibrium between 100 and 102.
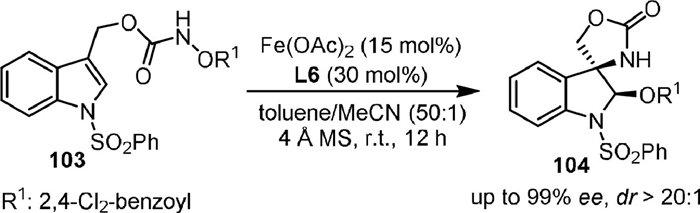
Later, the enantioselective intramolecular aminohydroxylation of indolyl-substituted carbamate (103) was developed under the catalysis of Fe(OAc)2-L6 (Scheme 22), from which a series of biologically active asymmetric 3-amino oxindoles and 3-amino indolanes could be synthesized in 3 steps [92].

|
Download:
|
| Scheme 22. Iron(Ⅱ)-catalyzed intramolecular aminohydroxylation of indoles. | |
{kind=link}
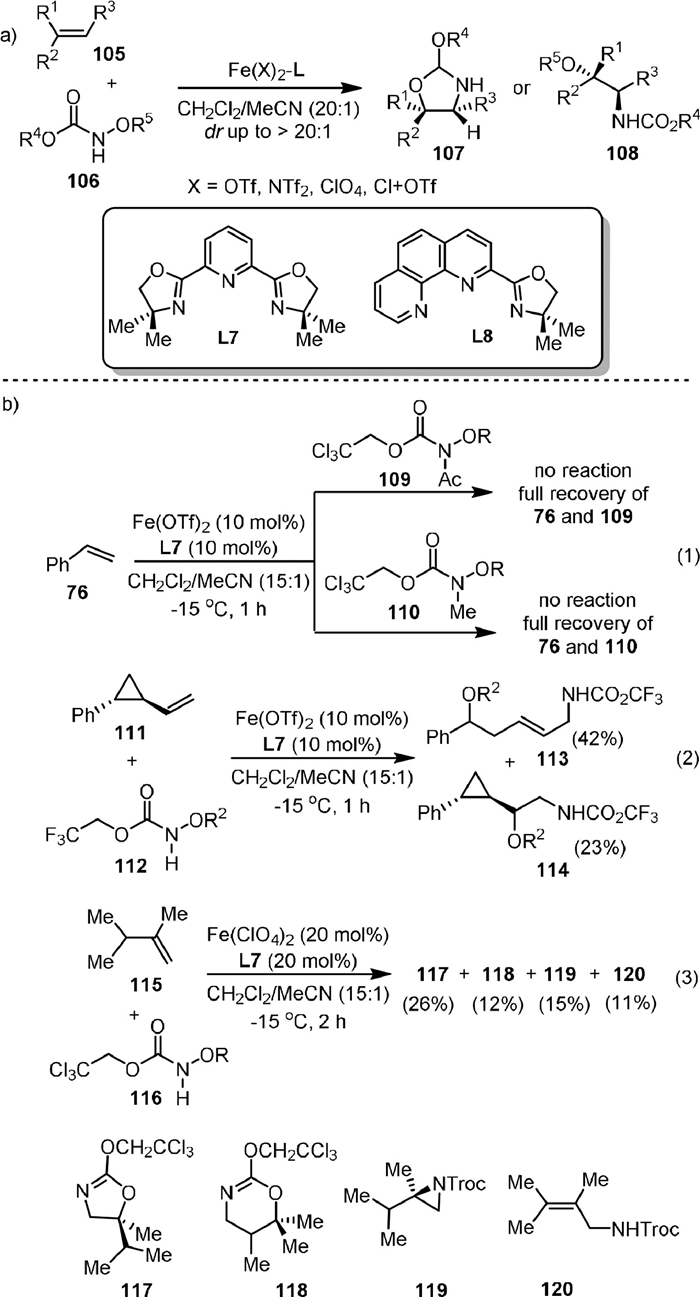
In 2014, the Xu group [93] reported the intermolecular amino-oxygenation of olefins in a diastereoselective manner with bench-stable hydroxylamines 106 as both nitrene precursor and oxidant (Scheme 23a). The method is applicable to a broad range of synthetically valuable alkenes that are not compatible with previously reported amino-oxygenation methods. It was found that the achiral bisoxazoline PyBOX ligand L7 was uniquely effective for most of the substrates, while L8 was more efficient for some conjugated dienes. Three control experiments were carried out to gain insight into the reaction mechanism (Scheme 23b). The hydroxylamines (109, 110) without the N−H moiety failed to react with the olefin, indicating the N−H group was critical for the reaction (Eq. 1). Radical clock experiments indicated that the reaction involved a stepwise process in which a radical amination step is involved (Eq. 2). The reaction of isopropyl-substituted terminal olefin (111) with hydroxylamine gave four products, including the standard 1,2-amino-oxygenation product 117, 1,3-amino-oxygenation product 118, aziridine 119, and allylic amine 120, indicating that the carbocation was involved in the reaction (Eq. 3). In addition, the conversion of the product 117 to other products did not occur under the reaction conditions, suggesting that the aziridine might not be an intermediate in the reaction. Later, they described the scope and limitations of the reaction and reported four practical procedures for the gram-scale aminohydroxylation of styrene, glycals, and indene [94].

|
Download:
|
| Scheme 23. Iron-catalyzed asymmetric olefin amino-oxygenation and control experiments to probe reaction mechanisms. | |
{kind=link}
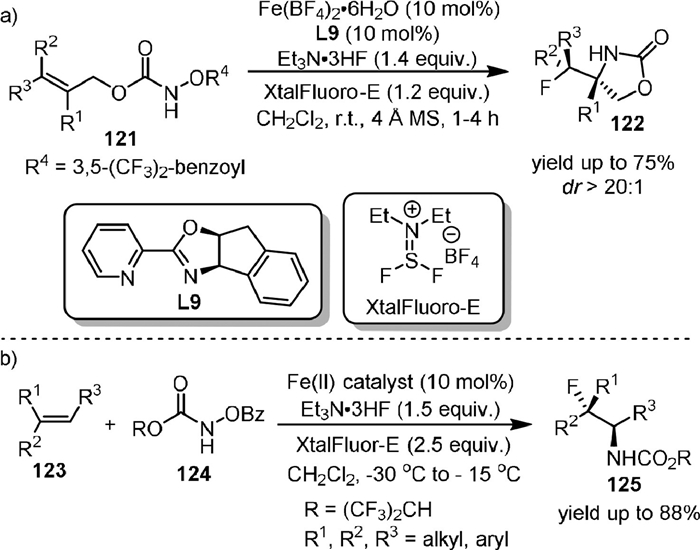
In the same year, they [95] reported the iron(Ⅱ)-catalyzed intramolecular diastereoselective aminofluorination of hydroxylamines (121) with Et3N·3HF as the fluorine source (Scheme 24a). The resulting fluoro oxazolidinones (122) could be conveniently transformed to β-fluoro primary amines and amino acids after further derivatization. It was found that the complex of Fe(BF4)2 and hybrid pyridine-oxazoline ligand L9 was the optimal catalyst and the XtalFluor-E could effectively minimize the competing aminohydroxylation process by acting as a carboxylate trapping reagent.

|
Download:
|
| Scheme 24. Iron-catalyzed olefin aminofluorination. | |
{kind=link}
Based on the above work, they developed the intermolecular aminofluorination of olefins in 2016 (Scheme 24b) [96]. A broad range of unfunctionalized olefins including nonstyrenyl olefins could be converted to synthetically valuable internal vicinal fluoro carbamates (125) with high regioselectivity. After rigorous screening, the crystalline solid Fe(L7)-(MeCN)(H2O)2(BF4)2 was found to be effective for this reaction. Preliminary mechanistic studies suggested that an asymmetric version of intermolecular olefins aminofluorination could be achieved when using chiral iron catalysts, and both an iron-nitrenoid and carbocation species might be involved in this transformation as key intermediates.
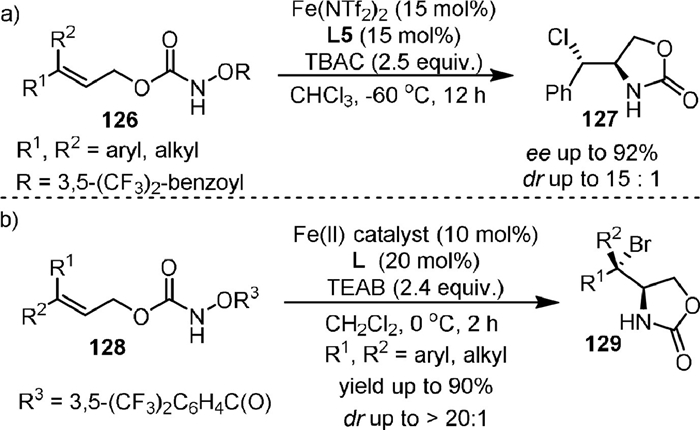
In 2015, they [97] reported the Fe(Ⅱ)-catalyzed intramolecular enantioselective and diastereoselective aminochlorination of allylic carbamates 126 (Scheme 25a), affording amino chlorides with contiguous stereogenic centers. Tetra-n-butylammonium chloride (TBAC) provided the chloride ion for this transformation. The method is applicable to a broad range of synthetically valuable internal olefins that are not compatible with other methods for asymmetric olefin aminochlorination. Preliminary mechanistic studies suggested that an iron-nitrenoid intermediate derived from FeCl2 might be involved in the reaction and the stereoselective chlorine atom-transfer might be related to the cleavage of the Fe-Cl bond. Similarly, the diastereoselective aminobromination of both internal and terminal olefins using iron(Ⅱ) catalyst was disclosed (Scheme 25b) [98]. The hydroxylamine (128) and tetraethylammonium bromide (TEAB) provided nitrogen and bromine for the reaction, respectively. It was found that both the diastereoselectivity and enantioselectivity of the transformation could be controlled by the iron-ligand complex.

|
Download:
|
| Scheme 25. Iron-catalyzed olefin aminochlorination and aminobromination. | |
{kind=link}
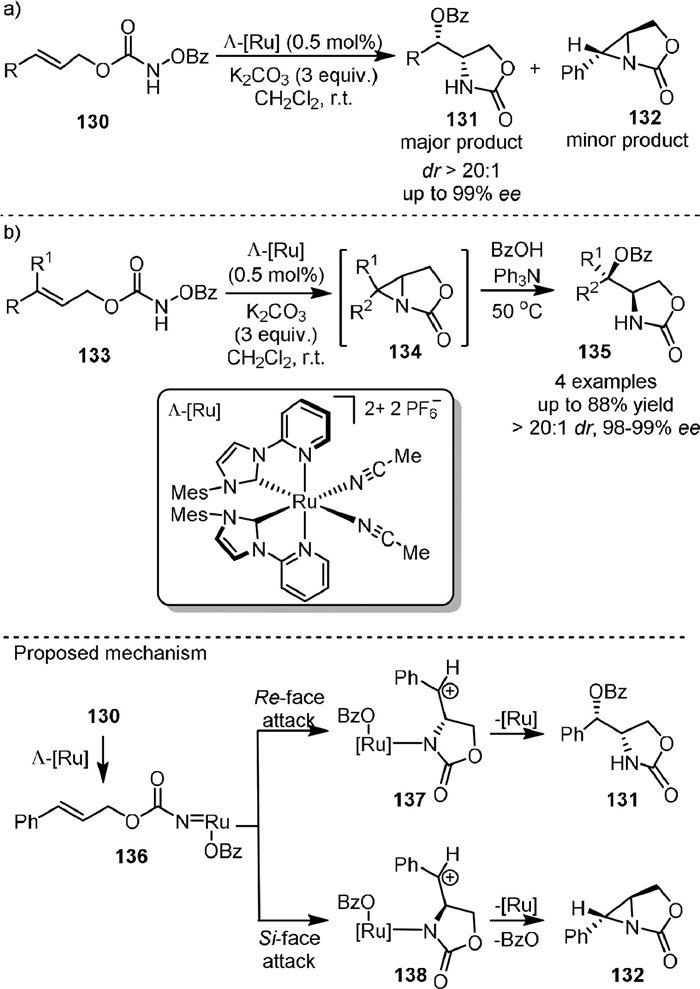
In 2020, Meggers [99] reported the asymmetric ring-closing aminooxygenation of 1,2-disubstituted alkenes (130) with N-benzoyloxycarbamates using the chiral-at-metal ruthenium NHC catalyst Λ-[Ru] (Scheme 26a). The reaction proceeded with excellent diastereoselectivity (typically > 20:1 dr) and enantioselectivities (up to 99% ee). However, the trisubstituted alkenes (133) were exclusively transformed to unstable aziridines (134) under the standard reaction conditions (Scheme 26b). The product 134 underwent ring-opening reactions with benzoic acid in the presence of Ph3N, affording aminooxygenation products (135) as single diastereomers. The proposed mechanism suggested that 130 reacted with Λ-[Ru] to form the ruthenium nitrenoid intermediate by migration of the OBz group (136), and then carbocation intermediates (137 and 138) with opposite configurations could be generated from the addition of nitrene to the Re-face and Si-face of the prochiral alkene, respectively. 137 could lead to the desired product 131, while 138 would form aziridine due to steric and/or stereoelectronic reasons.

|
Download:
|
| Scheme 26. Rutheuium-catalyzed alkene aminooxygenation. | |
{kind=link}
Compared with the interesting reactions for the bifunctionalization of alkenes, the reactions with alkynes have been much less investigated. Only one example was known recently, which leads to the carboxyamidation products by activation of alkyne-tethered N-benzoyloxycarbamates with PtCl2/CO or FeCl3, as reported by Lee and coworkers [100].
6. Other nitrene transfer reactions of carbamatesIn addition to the above-mentioned reactions, there are other types of nitrene transfer reactions of carbamates, through which N−S, C−N and N−N bonds are formed, thereby leading to complex molecular frameworks.
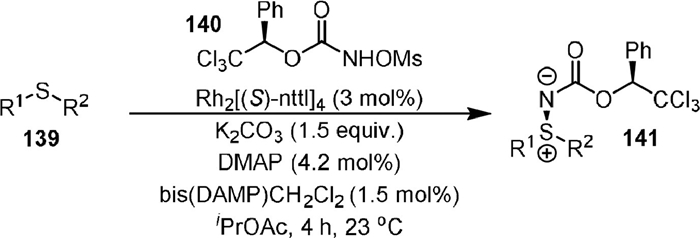
In 2014, Lebel [101] reported the stereoselective amination of thioethers (139) with chiral N-mesyloxycarbamates (140) to produce chiral sulfilimines (141, Scheme 27) in high yields and diastereomeric ratios. The chiral dirhodium(Ⅱ) carboxylate Rh[(S)-ntll]4 was used as the catalyst for the reaction. It was found that the diastereomeric ratio of the product could be significantly increased by the addition of 4-dimethylaminopyridine (DMAP) and bis(DMAP)CH2Cl2. The UV-vis spectra indicated that a thioether-Rh-DMAP complex was involved in this reaction, and an X-ray crystal structure of the (DMAP)2·[Rh24] complex was obtained. The cyclic voltammetry experiments indicated that RhⅡ-RhⅢ complex might be the catalytically active species.

|
Download:
|
| Scheme 27. Rhodium-catalyzed stereoselective amination of and thioethers. | |
{kind=link}
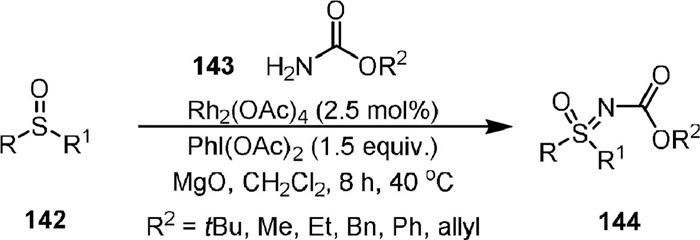
In 2015, Luisi and Bull [102] disclosed the synthesis of N-protected sulfoximines (144) by Rh(Ⅱ)-catalyzed intermolecular amination of sulfoxides (142) with carbamates under mild conditions (Scheme 28). A range of sulfoximine carbamates including 4-membered thietaneoximines and derivatives containing Boc, Cbz, methyl, ethyl, phenyl, and allyl groups could be obtained by the method. The N-Boc and N-Cbz protected sulfoximines could undergo Suzuki cross-couplings reaction, and could be easily deprotected to form free NH-sulfoximines [103,104].

|
Download:
|
| Scheme 28. Rhodium-catalyzed intermolecular amination of sulfoxides. | |
{kind=link}
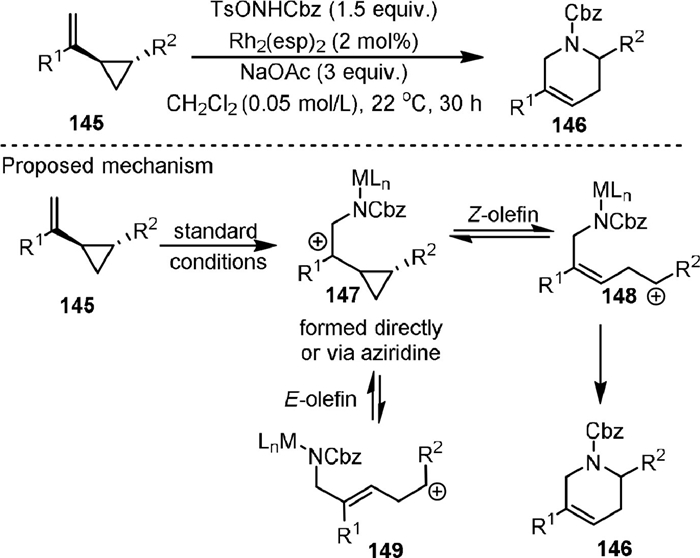
In 2019, Hilinski [105] reported the synthesis of six-membered nitrogen-containing heterocycles tetrahydropyridines (146) by the Rh(Ⅱ)-catalyzed [5 + 1] cycloaddition of aryl-substituted vinylcyclopropanes (145) with benzyl tosyloxycarbamate with high regioselectivity (Scheme 29). The product 146 could be further transformed regio- and stereoselectively to substituted piperidines, which were the most common N-heterocycles in FDA-approved drugs. Mechanistic studies of the reaction suggested that a stepwise cationic pathway was involved. The intermediate 147 could be generated under the reaction conditions, which was in equilibrium with the dipolar ring-opened geometric isomers 148 and 149, and the former one could lead to the final product.

|
Download:
|
| Scheme 29. Rhodium-catalyzed [5 + 1] cycloaddition of vinylcyclopropane. | |
{kind=link}
While the C−H insertion reaction of metal-nitrene was widely pursued, a rare example of C−N bond insertion was reported by Nemoto in 2019 [106], leading to the efficient synthesis of unique diazacyclic systems with an N−N bond linkage (Scheme 30). Under the catalysis of [Rh2(esp-OMe)2], 151 was obtained as the major product while the C−H insertion product 152 was only formed in a small amount with the 151/152 ratio over 97:3. The selectivity is different from the reaction under silver catalysis reported previously by the same group [60]. Radical pathway was excluded by performing the reaction in the presence of radical-trapping agents and by the radical clock experiments. Experimental and theoretical analysis based on DFT were carried out to understand the C−H insertion process and to rationalize the origin of the chemoselectivity. It was proposed that in the amide insertion process, the intramolecular reaction of the nitrene and amide nitrogen formed a rhodium-coordinated N−N ylide intermediate (154), which underwent the [1,2] acyl group migration to form the final product 151. Although this process is thermodynamically unfavorable, it is kinetically faster than the competing C−H insertion reaction. The insertion of Rh-nitrene into sulfonamide S−N bonds could also be achieved.

|
Download:
|
| Scheme 30. Ylide formation with amide followed by the acyl transfer. | |
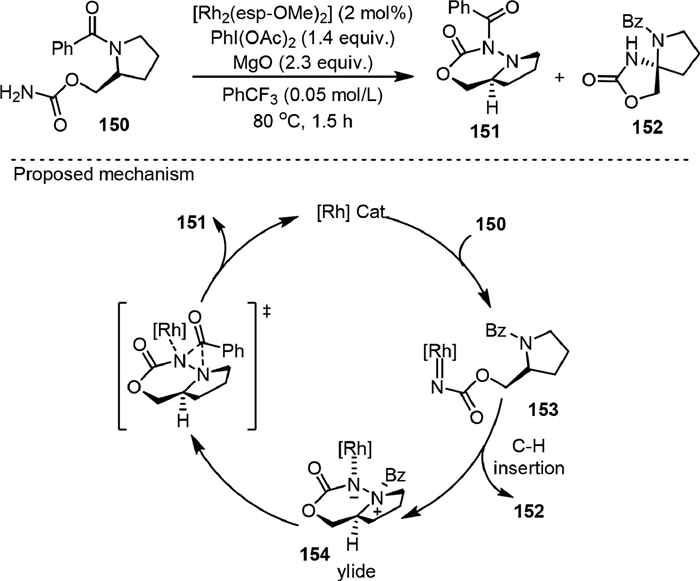{kind=link}
In 2020, Ellman [107] developed a route to [5,6]- and [6,6]-bicyclic [1,3,5]-triazinones (157) via the carbamylation of various imines (155) with ethyl (pivaloyloxy)carbamate (156) and subsequent cyclization under the catalysis of Rh(Ⅲ) (Scheme 31a). A broad scope of N-heterocycles could be successfully incorporated into the bicyclic skeleton, and the R substituent on the triazinone ring could be aliphatic, aromatic and alkoxy groups. Notably, the efficiency of this method could be further improved in a one-pot three-component procedure, in which the imines were in situ generated from aldehydes (158) and amino N-heterocycles (159) (Scheme 31b).

|
Download:
|
| Scheme 31. Rhodium-catalyzed synthesis of [5,6]- and [6,6]-bicyclic [1,3,5]-triazinones. | |
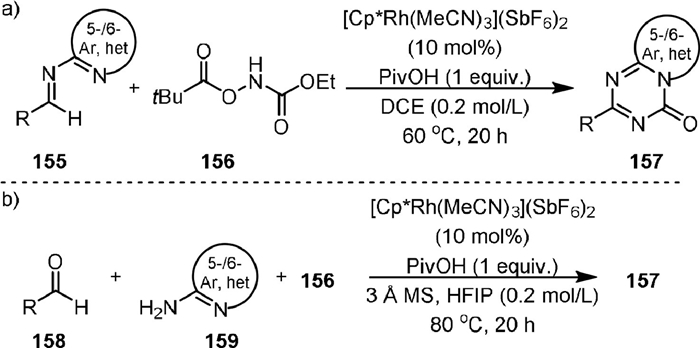{kind=link}
7. Summary and outlook
The recent development of transition metal-catalyzed nitrene transfer reactions with carbamates is briefly described in this review. The carbamate precursors are readily available and bench stable and their nitrene transfer reactions involving N−O bond cleavage could generally occur under mild conditions with a proper catalyst. While the Rh(Ⅱ) complexes were conventionally the most prevalent catalyst to form the Rh(Ⅱ)-nitrenoid intermediate, more and more examples have been disclosed by using cheaper catalytic systems involving complexes of Fe, Ru, Ag, Cu, Co, and others. The in-situ formed metal-nitrenoid intermediates underwent important transformations including C−H bond amination, aziridination, bifunctionalization of alkenes and others, from which the synthesis of biologically active nitrogen-containing compounds with high enantioselectivity were reported, showing the effectiveness and usefulness of carbamates as the nitrene precursors. In particular, the intramolecular sp3C−H aminations are remarkable for facile access to oxazolidinones, which have been found to be a new class of synthetic antibacterials with activity against Gram-positive bacteria and anaerobic bacteria [108].
While great progress has been achieved in the field, future attention may be paid to the following aspects in view of green and sustainable transformations. First, compared with the high atomic economy in nitrene transfers with azide derivatives and 1,4,2-dioxazol-5-ones as precursors, in which only N2 and CO2 are released as waste, the formation of organic by-products are inevitable in the reactions with carbamates. Second, the cheaper catalysts such as the iron complexes were found to be useful for the bifunctionalization of alkenes, however, the using of inexpensive metals for more types of nitrene transfer reactions with carbamates remains beyond reach. The last but not the least, the carbamate functionality is contained in the nitrene transfer products and its removal generally requires strong basic conditions, which should limit the functional group tolerance for amine formation. It is anticipated that the insights provided in this review will be beneficial for eliciting further development in the field.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the financial support provided by the National Natural Science Foundation of China (Nos. 21572163 and 21873074) and the Wenzhou Science & Technology Bureau (No. G20210032).
| [1] |
H.M.L. Davies, J.R. Manning, Nature 451 (2008) 417-424. DOI:10.1038/nature06485 |
| [2] |
G. Dequirez, V. Pons, P. Dauban, Angew. Chem. Int. Ed. 51 (2012) 7384-7395. DOI:10.1002/anie.201201945 |
| [3] |
J.L. Roizen, M.E. Harvey, J. Du Bois, Acc. Chem. Res. 45 (2012) 911-922. DOI:10.1021/ar200318q |
| [4] |
Y. Park, Y. Kim, S. Chang, Chem. Rev. 117 (2017) 9247-9301. DOI:10.1021/acs.chemrev.6b00644 |
| [5] |
F.A.H. Nasab, L.Z. Fekri, A. Monfared, et al., RSC Adv. 8 (2018) 18456-18469. DOI:10.1039/C8RA00356D |
| [6] |
H. Zhang, H. Wang, Y. Jiang, et al., Chem. Eur. J. 26 (2020) 17289-17317. DOI:10.1002/chem.202001414 |
| [7] |
K. Peng, Z.B. Dong, Eur. J. Org. Chem. 2020 (2020) 5488-5495. DOI:10.1002/ejoc.202000541 |
| [8] |
K. Omura, T. Uchida, R. Irie, T. Katsuki, Chem. Commun. (2004) 2060-2061. |
| [9] |
B.J. Stokes, H. Dong, B.E. Leslie, et al., J. Am. Chem. Soc. 129 (2007) 7500-7501. DOI:10.1021/ja072219k |
| [10] |
M. Shen, B.E. Leslie, T.G. Driver, Angew. Chem. Int. Ed. 47 (2008) 5056-5059. DOI:10.1002/anie.200800689 |
| [11] |
B.J. Stokes, K.J. Richert, T.G. Driver, J. Org. Chem. 74 (2009) 6442-6451. DOI:10.1021/jo901224k |
| [12] |
Y. Feng, Z. Zhang, Q. Fu, et al., Chin. Chem. Lett. 31 (2020) 58-60. DOI:10.1016/j.cclet.2019.05.013 |
| [13] |
T. Li, H. Yan, X. Li, et al., J. Org. Chem. 81 (2016) 12031-12037. DOI:10.1021/acs.joc.6b02322 |
| [14] |
W. Zhou, M. Zhang, H. Li, et al., Org. Lett. 19 (2017) 10-13. DOI:10.1021/acs.orglett.6b02850 |
| [15] |
M.N. Zhao, Z.H. Ren, D.S. Yang, et al., Org. Lett. 20 (2018) 1287-1290. DOI:10.1021/acs.orglett.7b04007 |
| [16] |
A. Prechter, G. Henrion, P. Faudot dit Bel, et al., Angew. Chem. Int. Ed. 53 (2014) 4959-4963. DOI:10.1002/anie.201402470 |
| [17] |
Y. Huang, C. Pi, Z. Tang, et al., Chin. Chem. Lett. 31 (2020) 3237-3240. DOI:10.1016/j.cclet.2020.08.046 |
| [18] |
C.L. Zhong, B.Y. Tang, P. Yin, et al., J. Org. Chem. 77 (2012) 4271-4277. DOI:10.1021/jo202663n |
| [19] |
Y. Park, K.T. Park, J.G. Kim, et al., J. Am. Chem. Soc. 137 (2015) 4534-4542. DOI:10.1021/jacs.5b01324 |
| [20] |
Y. Yu, Z. Xia, Q. Wu, et al., Chin. Chem. Lett. 32 (2021) 1263-1266. DOI:10.1016/j.cclet.2020.09.020 |
| [21] |
J.L. Liang, J.S. Huang, X.Q. Yu, et al., Chem. Eur. J. 8 (2002) 1563-1572. DOI:10.1002/1521-3765(20020402)8:7<1563::AID-CHEM1563>3.0.CO;2-V |
| [22] |
M. Anada, M. Tanaka, N. Shimada, et al., Tetrahedron 65 (2009) 3069-3077. DOI:10.1016/j.tet.2008.10.091 |
| [23] |
S. Lin, B. Lin, Z. Zhang, et al., Org. Lett. 24 (2022) 3302-3306. DOI:10.1021/acs.orglett.2c01238 |
| [24] |
T. Qi, N. Fang, W. Huang, et al., Org. Lett. 24 (2022) 5674-5678. DOI:10.1021/acs.orglett.2c01990 |
| [25] |
X. Zhang, B. Lin, J. Chen, et al., Org. Lett. 23 (2021) 819-825. DOI:10.1021/acs.orglett.0c04043 |
| [26] |
M. Hou, Z. Zhang, X. Lai, et al., Org. Lett. 24 (2022) 4114-4118. DOI:10.1021/acs.orglett.2c01176 |
| [27] |
J. Li, Y. Wang, H. Xie, et al., Mol. Catal. 516 (2021) 111993. DOI:10.1016/j.mcat.2021.111993 |
| [28] |
L. Xu, Q. Zhu, G. Huang, et al., J. Org. Chem. 77 (2012) 3017-3024. DOI:10.1021/jo202431q |
| [29] |
T. Zhou, W. Guo, Y. Xia, Chem. Eur. J. 21 (2015) 9209-9218. DOI:10.1002/chem.201500558 |
| [30] |
W. Guo, T. Zhou, Y. Xia, Organometallics 34 (2015) 3012-3020. DOI:10.1021/acs.organomet.5b00317 |
| [31] |
W. Guo, Y. Xia, J. Org. Chem. 80 (2015) 8113-8121. DOI:10.1021/acs.joc.5b01201 |
| [32] |
J. Chen, W. Guo, Y. Xia, J. Org. Chem. 81 (2016) 2635-2638. DOI:10.1021/acs.joc.6b00003 |
| [33] |
S.V. Athavale, S. Gao, Z. Liu, S.C. Mallojjala, et al., Angew. Chem. Int. Ed. 60 (2021) 24864-24869. DOI:10.1002/anie.202110873 |
| [34] |
T. Katsuki, Chem. Lett. 34 (2005) 1304-1309. DOI:10.1246/cl.2005.1304 |
| [35] |
T. Uchida, T. Katsuki, Chem. Rec. 14 (2014) 117-129. DOI:10.1002/tcr.201300027 |
| [36] |
A.K. Ghosh, M. Brindisi, A. Sarkar, ChemMedChem 13 (2018) 2351-2373. DOI:10.1002/cmdc.201800518 |
| [37] |
H. Hayashi, T. Uchida, Eur. J. Org. Chem. 2020 (2020) 909-916. DOI:10.1002/ejoc.201901562 |
| [38] |
T. Shimbayashi, K. Sasakura, A. Eguchi, et al., Chem. Eur. J. 25 (2019) 3156-3180. DOI:10.1002/chem.201803716 |
| [39] |
K.M. van Vliet, B. de Bruin, ACS Catal. 10 (2020) 4751-4769. DOI:10.1021/acscatal.0c00961 |
| [40] |
Y. Tamura, T. Uchida, T. Katsuki, Tetrahedron Lett. 44 (2003) 3301-3303. DOI:10.1016/S0040-4039(03)00609-9 |
| [41] |
X. Dong, M. Shang, S. Chen, et al., J. Org. Chem. 87 (2022) 13990-14004. DOI:10.1021/acs.joc.2c01636 |
| [42] |
S.Y. Moon, U.B. Kim, D.B. Sung, et al., J. Org. Chem. 80 (2015) 1856-1865. DOI:10.1021/jo502828r |
| [43] |
T. Bach, B. Schlummer, K. Harms, Chem. Commun. (2000) 287-288. |
| [44] |
M. Murakami, T. Uchida, T. Katsuki, Tetrahedron Lett. 42 (2001) 7071-7074. DOI:10.1016/S0040-4039(01)01448-4 |
| [45] |
H.M.L. Davies, M.S. Long, Angew. Chem. Int. Ed. 44 (2005) 3518-3520. DOI:10.1002/anie.200500554 |
| [46] |
P. Müller, C. Fruit, Chem. Rev. 103 (2003) 2905-2920. DOI:10.1021/cr020043t |
| [47] |
P. Ruiz-Castillo, S.L. Buchwald, Chem. Rev. 116 (2016) 12564-12649. DOI:10.1021/acs.chemrev.6b00512 |
| [48] |
C.G. Espino, Bois J.Du, Angew. Chem. Int. Ed. 40 (2001) 598-600. DOI:10.1002/1521-3773(20010202)40:3<598::AID-ANIE598>3.0.CO;2-9 |
| [49] |
A. Hinman, Bois J.Du, J. Am. Chem. Soc. 125 (2003) 11510-11511. DOI:10.1021/ja0368305 |
| [50] |
X. Lin, C. Zhao, C.M. Che, et al., Chem. Asian J. 2 (2007) 1101-1108. DOI:10.1002/asia.200700068 |
| [51] |
K.A. Parker, W. Chang, Org. Lett. 7 (2005) 1785-1788. DOI:10.1021/ol050356l |
| [52] |
R.D. Grigg, J.W. Rigoli, S.D. Pearce, et al., Org. Lett. 14 (2012) 280-283. DOI:10.1021/ol203055v |
| [53] |
K.M. Brummond, B. Mitasev, Org. Lett. 6 (2004) 2245-2248. DOI:10.1021/ol0492391 |
| [54] |
H. Lebel, K. Huard, S. Lectard, J. Am. Chem. Soc. 127 (2005) 14198-14199. DOI:10.1021/ja0552850 |
| [55] |
K. Huard, H. Lebel, Chem. Eur. J. 14 (2008) 6222-6230. DOI:10.1002/chem.200702027 |
| [56] |
T. Yakura, Y. Yoshimoto, C. Ishida, Chem. Pharm. Bull. 55 (2007) 1385-1389. DOI:10.1248/cpb.55.1385 |
| [57] |
R.P. Reddy, H.M.L. Davies, Org. Lett. 8 (2006) 5013-5016. DOI:10.1021/ol061742l |
| [58] |
Y. Cui, C. He, Angew. Chem. Int. Ed. 43 (2004) 4210-4212. DOI:10.1002/anie.200454243 |
| [59] |
M. Ju, M. Huang, L.E. Vine, et al., Nature Catal. 2 (2019) 899-908. DOI:10.1038/s41929-019-0339-y |
| [60] |
S. Harada, M. Kobayashi, M. Kono, et al., ACS Catal. 10 (2020) 13296-13304. DOI:10.1021/acscatal.0c04057 |
| [61] |
M. Ju, E.E. Zerull, J.M. Roberts, et al., J. Am. Chem. Soc. 142 (2020) 12930-12936. DOI:10.1021/jacs.0c05726 |
| [62] |
H. Jung, H. Keum, J. Kweon, et al., J. Am. Chem. Soc. 142 (2020) 5811-5818. DOI:10.1021/jacs.0c00868 |
| [63] |
Y. Tan, S. Chen, Z. Zhou, et al., Angew. Chem. Int. Ed. 59 (2020) 21706-21710. DOI:10.1002/anie.202009335 |
| [64] |
H. Lebel, K. Huard, Org. Lett. 9 (2007) 639-642. DOI:10.1021/ol062953t |
| [65] |
H. Lebel, C. Spitz, O. Leogane, et al., Org. Lett. 13 (2011) 5460-5463. DOI:10.1021/ol2021516 |
| [66] |
J. Jeong, H. Jung, D. Kim, et al., ACS Catal. 12 (2022) 8127-8138. DOI:10.1021/acscatal.2c02612 |
| [67] |
F. Collet, R.H. Dodd, P. Dauban, Chem. Commun. (2009) 5061-50742009. |
| [68] |
K.H. Ng, A.S.C. Chan, W.Y. Yu, J. Am. Chem. Soc. 132 (2010) 12862-12864. DOI:10.1021/ja106364r |
| [69] |
K.H. Ng, F.N. Ng, W.Y. Yu, Chem. Commun. 48 (2012) 11680-11682. DOI:10.1039/c2cc36502b |
| [70] |
R. Giri, J.Q. Yu, J. Am. Chem. Soc. 30 (2008) 114082-114083. |
| [71] |
C. Grohmann, H. Wang, F. Glorius, Org. Lett. 15 (2013) 3014-3017. DOI:10.1021/ol401209f |
| [72] |
B. Zhou, J. Du, Y. Yang, et al., Org. Lett. 16 (2014) 592-595. DOI:10.1021/ol403477w |
| [73] |
P. Patel, S. Chang, Org. Lett. 16 (2014) 3328-3331. DOI:10.1021/ol501338h |
| [74] |
P. Patel, S. Chang, ACS Catal. 5 (2015) 853-858. DOI:10.1021/cs501860b |
| [75] |
R. Singh, K. Nagesh, M. Parameshwar, ACS Catal. 6 (2016) 6520-6524. DOI:10.1021/acscatal.6b02237 |
| [76] |
K. Arai, Y. Ueda, K. Morisaki, et al., Chem. Commun. 54 (2018) 2264-2267. DOI:10.1039/C7CC09952E |
| [77] |
L. Degennaro, P. Trinchera, R. Luisi, Chem. Rev. 114 (2014) 7881-7929. DOI:10.1021/cr400553c |
| [78] |
Y. Zhu, Q. Wang, R.G. Cornwall, et al., Chem. Rev. 114 (2014) 8199-8256. DOI:10.1021/cr500064w |
| [79] |
C.J. Hayes, P.W. Beavis, L.A. Humphries, Chem. Commun. (2006) 4501-45022006. |
| [80] |
R. Liu, S.R. Herron, S.A. Fleming, J. Org. Chem. 72 (2007) 5587-5591. DOI:10.1021/jo0705014 |
| [81] |
H. Lebel, S. Lectard, M. Parmentier, Org. Lett. 9 (2007) 4797-4800. DOI:10.1021/ol702152e |
| [82] |
M. Barani, S. Fioravanti, L. Pellacani, et al., Tetrahedron 50 (1994) 11235-11238. DOI:10.1016/S0040-4020(01)89425-4 |
| [83] |
S. Fioravanti, G. Luna, L. Pellacani, et al., Tetrahedron 53 (1997) 4779-4786. DOI:10.1016/S0040-4020(97)00161-0 |
| [84] |
H. Lebel, M. Parmentier, Pure Appl. Chem. 82 (2010) 1827-1833. DOI:10.1351/PAC-CON-09-12-12 |
| [85] |
A. Padwa, T. Stengel, Org. Lett. 4 (2002) 2137-2139. DOI:10.1021/ol0259490 |
| [86] |
A. Padwa, A.C. Flick, C.A. Leverettl, et al., J. Org. Chem. 69 (2004) 6377-6386. DOI:10.1021/jo048990k |
| [87] |
E. Levites-Agababa, E. Menhaji, L.N. Perlson, et al., Org. Lett. 4 (2002) 863-865. DOI:10.1021/ol025634k |
| [88] |
R. Gupta, K.M. Sogi, S.E. Bernard, et al., Org. Lett. 11 (2009) 1527-1530. DOI:10.1021/ol900126q |
| [89] |
W.P. Unsworth, S.G. Lamont, J. Robertson, Tetrahedron 70 (2014) 7388-7394. DOI:10.1016/j.tet.2014.06.051 |
| [90] |
A.C. Willis Masruri, M.D. McLeod, J. Org. Chem. 77 (2012) 8480-8491. DOI:10.1021/jo301372y |
| [91] |
G.S. Liu, Y.Q. Zhang, Y.A. Yuan, et al., J. Am. Chem. Soc. 135 (2013) 3343-3346. DOI:10.1021/ja311923z |
| [92] |
Y.Q. Zhang, Y.A. Yuan, G.S. Liu, et al., Org. Lett. 15 (2013) 3910-3913. DOI:10.1021/ol401666e |
| [93] |
D.F. Lu, C.L. Zhu, Z.X. Jia, et al., J. Am. Chem. Soc. 136 (2014) 13186-13189. DOI:10.1021/ja508057u |
| [94] |
C.L. Zhu, D.F. Lu, J.D. Sears, et al., Synthesis 48 (2016) 3031-3041. DOI:10.1055/s-0035-1562515 |
| [95] |
D.F. Lu, G.S. Liu, C.L. Zhu, et al., Org. Lett. 16 (2014) 2912-2915. DOI:10.1021/ol501051p |
| [96] |
D.F. Lu, C.L. Zhu, J.D. Sears, et al., J. Am. Chem. Soc. 138 (2016) 11360-11367. DOI:10.1021/jacs.6b07221 |
| [97] |
C.L. Zhu, J.S. Tian, Z.Y. Gu, et al., Chem. Sci. 6 (2015) 3044-3050. DOI:10.1039/C5SC00221D |
| [98] |
J.S. Tian, C.L. Zhu, Y.R. Chen, et al., Synthesis 47 (2015) 1709-1715. DOI:10.1055/s-0034-1378719 |
| [99] |
Y. Tan, F. Han, M. Hemming, et al., Org. Lett. 22 (2020) 6653-6656. DOI:10.1021/acs.orglett.0c02452 |
| [100] |
S. Su, T. Wu, Y. Xia, et al., Chem. Eur. J. 29 (2023) e202203371. DOI:10.1002/chem.202203371 |
| [101] |
H. Lebel, H. Piras, J. Bartholoméüs, Angew. Chem. Int. Ed. 53 (2014) 7300-7304. DOI:10.1002/anie.201402961 |
| [102] |
M. Zenzola, R. Doran, R. Luisi, et al., J. Org. Chem. 80 (2015) 6391-6399. DOI:10.1021/acs.joc.5b00844 |
| [103] |
T.E. Barder, S.D. Walker, J.R. Martinelli, et al., J. Am. Chem. Soc. 127 (2005) 4685-4696. DOI:10.1021/ja042491j |
| [104] |
A. Fürstner, A. Leitner, Angew. Chem. Int. Ed. 41 (2002) 609-612. DOI:10.1002/1521-3773(20020215)41:4<609::AID-ANIE609>3.0.CO;2-M |
| [105] |
L.A. Combee, S.L. Johnson, J.E. Laudenschlager, et al., Org. Lett. 21 (2019) 2307-2311. DOI:10.1021/acs.orglett.9b00594 |
| [106] |
M. Kono, S. Harada, T. Nemoto, Chem. Eur. J. 25 (2019) 3119-3124. DOI:10.1002/chem.201805878 |
| [107] |
D.N. Confair, N.S. Greenwood, B.Q. Mercado, et al., Org. Lett. 22 (2020) 8993-8997. DOI:10.1021/acs.orglett.0c03393 |
| [108] |
N. Selvakumar, D. Srinivas, M.K. Khera, et al., J. Med. Chem. 45 (2002) 3953-3962. DOI:10.1021/jm020092y |