 2024, Vol. 35
2024, Vol. 35 


b College of Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center (RECAST), Frontiers Science Center for New Organic Matter, Nankai University, Tianjin 300071, China
Organic electrochemistry provides a novel and powerful strategy for organic synthesis that uses "greener" electricity instead of traditional chemical redox reagents [1–13]. Electrochemical transformations have been widely used in various research areas, such as alkene difunctionalizations [14–22], C–H oxidations [23–28], C–H activations [29–34], reduction reactions [35–39] among others [40–47]. Electrochemical organic synthesis has also shown its potential for benzylic C–H oxidation through direct [48–53] or mediated electro-oxidation (mainly focused on organic mediators) [54–58] processes, being efficient and environmentally friendly. However, there were still some issues lying in the way of putting this strategy to a wider application, such as limited functional group tolerance, sensitive group incompatibility, unsatisfactory selectivity, and most strategies exploit solely oxidative half-cell reaction. Due to the good modulation of the reaction selectivity and the high tolerance of functional groups, transition metal catalysis has emerged as one of the most important avenues for selectivity control in modern synthetic chemistry [59–64]. We envisioned that transition-metal catalysis could be used for improving the functional group tolerance in selective benzylic C–H oxidation of functionalized alkyl arenes. To the best of our knowledge, selective transition metal-catalyzed electrochemical benzylic C–H oxidation of functionalized alkyl arenes with sensitive functional groups has thus far proven elusive [65,66].
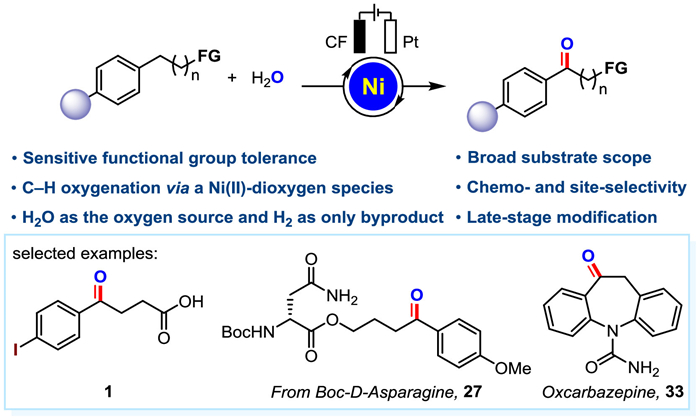
Following our continuous interest in sustainable electrochemical transformation [67–71], and also inspired by the recent elegant example of nickel-catalyzed electro-oxidation of sulfides [72], herein we report an unprecedented paired nickel-electrocatalyzed benzylic C–H oxygenation of functionalized alkyl arenes using water as an oxygen source (Fig. 1). Notably, various functionalized aromatic ketones, including examples with sensitive functional groups, underwent the transformation smoothly. For example, iodinated aromatics were well tolerated in this reaction. Moreover, aldehydes and alcohols, which can be easily over-oxidized, were also compatible in this paired nickel-catalyzed system. Notable features of this strategy include: (a) Excellent tolerance to a wide variety of sensitive functional groups, such as free carboxylic acids, amides, amino acids, esters, halogens, alcohols, and aldehydes; (b) High chemo- and site- selectivity; (c) Late-stage oxygenation of natural compounds and drug derivatives; (d) Short-lived reactive aryl radical cation intermediate was captured by the combination of a bipolar ultramicroelectrode (BUME) with nano-electrospray ionization mass spectrometry. Of particular note, this facile and selective paired nickel-catalyzed electro-oxidation strategy could also be applied to the synthesis of natural products.

|
Download:
|
| Fig. 1. Selective nickel-electrocatalyzed benzylic C–H oxygenation of diverse functionalized alkyl arenes. | |
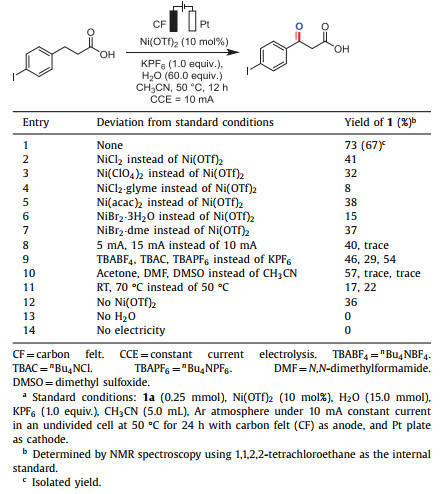
Initially, we employed 4-(4-iodophenyl)butanoic acid (1a) as the model substrate and investigated various reaction conditions with H2O under a constant current for the electrocatalytic oxidation reaction. After careful optimization, 4-(4-iodophenyl)-4-oxobutanoic acid (1) was obtained in 67% isolated yield, where the reaction was conducted in the presence of a catalytic amount of Ni(OTf)2 under a constant current (10 mA) in CH3CN with the addition of 60 equiv. of water, and KPF6 as the supporting electrolyte, carbon felt as the anode and platinum plate as the cathode (Table 1, entry 1). Other Ni(Ⅱ) salts with different counter ions, such as NiCl2, Ni(ClO4)2, Ni(acac)2, NiBr2·3H2O and NiBr2·dme were also tested and afforded the desired product in apparently lower yields (entries 2−7). Attempts to lower the current (5 mA) resulted in a significant decrease in efficiency (40% yield), and in contrast, no desired product was detected at 15 mA (entry 8). When TBABF4, TBAC and TBAPF6 were employed as the electrolytes in the reaction, it resulted in lower yields of the desired product (entry 9). Next, the effect of solvents, including N, N-dimethylformamide (DMF), acetone, and dimethyl sulfoxide (DMSO) was tested. As a result, CH3CN was found to be the best choice for this conversion (entry 10). Furthermore, a decrease in yield was observed when the reaction was carried out at room temperature or 70 ℃ (entry 11). Finally, control experiments confirmed the essential role of Ni(OTf)2, electricity and H2O, as no desired product could be detected in the absence of the latter two (entries 12−14, for more details, see Table S1 in Supporting information).
|
|
Table 1 Optimization of the reaction conditions. a |
{kind=link}
With the optimized reaction conditions in hand, we next investigated the scope and compatibility of the reaction (Scheme 1). To our delight, a set of representative functionalized alkyl arenes were proven to work smoothly in the reaction, affording the corresponding aryl ketone acids in moderate to excellent yields (1−6). After constant current electrolysis, the sensitive groups on substrates (precursors of 7−14) remained intact, and moderate to good yields of the desired products were obtained. The substrate with long alkyl chain bearing a terminal OTIPS group resulted in the deprotected hydroxyl product (15) in 78% yield. The benzyl electro-oxidation procedure could be extended to the late-stage functionalization of complex compounds. Alkylarenes derived from drugs, including picaridin, epiandrosterone, DL-isoborneol, esterone four, fenchol, fenbufen, retinoic acid receptor agonist derivatives, and DL-menthol, stearic acid, α-amino acid derivatives including N-acetyl-L-isoleucine, glutimicacid, Boc-L-proline and N-Boc-L-valine, Boc-D-asparagine, Boc-glycine were all amenable to the electro-oxidation transformation, providing the desired carbonyl compounds in moderate to good yields and diastereoselectivities 16−30 (17, 18, 23, 24, 25, and 27 were obtained as single diastereoisomer). Similarly, drug molecules fenofibrate (31), fenbufen (32) and oxcarbazepine (33) could be generated directly using our reaction conditions. These results indicated the synthetic potential of this method for the convenient introduction of carbonyl groups to complex compounds and drug molecules. Functionalized arenes with electron-withdrawing (34−39) or electron-donating (40−47) substituents all afforded the desired corresponding alkyl aryl ketones in moderate to good yields, and the sensitive functional groups remained untouched under the standard conditions. Similarly, the substrates with aryl substitution on the benzene ring also worked well under our reaction conditions and afforded the desired products in good yields (48−51). Notably, when substrate 52 bearing two benzylic positions was employed, the reaction occurred selectively at the benzylic position of the more electron-rich methoxy-substituted aryl group. We also observed that the oxidation reaction occurred preferentially on the ring when both a ring and alkyl chain reaction sites were available (54). When the benzylic position is linked to a ternary ring, the ternary ring did not undergo a ring-opening process after electrolysis and directly gave the product (53) in moderate yield. Simple 1-indanone (55) and benzohexacycles (56−58) all worked smoothly to generate the desired products. Interestingly, for diaryl-substituted substrates, the target molecules were obtained in excellent yields (59−74). For the substrate containing a methyl group on the aryl group, only one product was obtained and no aldehyde was detected, which might due to the difference in electron cloud density between the two reaction sites (63). The reaction also proceeded when substrates with heteroatomic rings were used, for example, the target product (71) was generated in 52% yield. Meanwhile, for the substrate 9,10-dihydroanthracene, the product anthraquinone (74) was obtained directly in one step under the standard reaction conditions, while no mono-oxidized product ketone was detected. It is noteworthy that there is no catalysis without Ni (more details are given in Fig. S1 in Supporting information).

|
Download:
|
| Scheme 1. Nickel-electrocatalyzed benzylic C–H oxygenation of functionalized alkyl arenes. a Standard conditions: arenes (0.25 mmol), Ni(OTf)2 (10 mol%), H2O (15.0 mmol), KPF6 (1.0 equiv.), CH3CN (5.0 mL), Ar atmosphere under 10 mA constant current in an undivided cell at 50 ℃ for 10−24 h, caron felt (CF) as anode, and Pt plate as cathode. b The reaction was conducted under a constant current of 5 mA for 12 h. c The starting material is triisopropyl(4-(4-methoxyphenyl)butoxy)silane. d The reaction was conducted under a constant current of 5 mA for 10 h. | |
{kind=link}
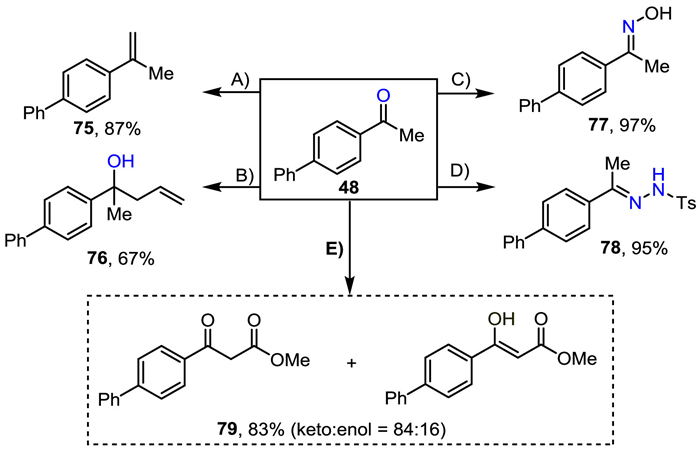
To further illustrate the synthetic practicability of this method, we carried out the product derivatizations (Scheme 2) [54,73,74]. 4-Acetylbiphenyl could be easily converted to olefin 75 in 87% yield by a Wittig reaction, which is a fundamental building block for bioactive molecules. Furthermore, 4-acetylbiphenyl could undergo a nucleophilic attack, furnishing the valuable alcohol 76 in one pot by adding allyl bromide and indium powder after the reaction was completed. In addition, 4-acetylbiphenyl could generate oxime 77 and hydrazone 78 in good yields. It could also react with dimethyl carbonate to form 79, which is an important drug precursor (more details are given in Supporting information).

|
Download:
|
| Scheme 2. Product derivatization. (A): 48 (1.0 equiv.), tBuOK (1.2 equiv.), PPh3CH3Br (2.4 equiv.), dry THF, 0 ℃, 1 h, then r.t., 5 h. (B): the first-step oxidation was conducted according to the general procedure then added allyl bromide (3.0 equiv.), In powder (3.0 equiv.), 55 ℃, 10 h. (C): 48 (1.0 equiv.), NH2OH·HCl (1.5 equiv.), NaOAc (2.6 equiv.), ethanol/water = 1/4, 95 ℃, 10 h. (D): 48 (1.0 equiv.), NH2NHTs (1.0 equiv.), dry methanol, 60 ℃, 1 h; (E): 48 (1.0 equiv.), NaH (3.0 equiv.), CO(OMe)2 (2.0 equiv.), toluene, reflux, 2 h. | |
{kind=link}
Next, we conducted some controlled experiments. First, reactions under the optimal conditions with different amounts of H2O were performed (Fig. S4 in Supporting information). Notably, the reaction did not show a steady upward trend when 10 and 30 equiv. of water were added, respectively, which might be due to the competition between the oxidation of substrate 48a and the oxygen evolution reaction (OER) of H2O (the oxidation potentials of 48a and H2O are 1.95 V vs. Ag/Ag+ and 1.91 V vs. Ag/Ag+, respectively). However, when we added 60, 80, and 100 equiv. of water, a gradual decrease in yield was observed, which might be due to the increasing oxygen production. We also conducted electricity on/off experiments, and the transformation was entirely suppressed in the absence of electricity (Table S3 in Supporting information), which confirmed the importance of the participation of electricity.
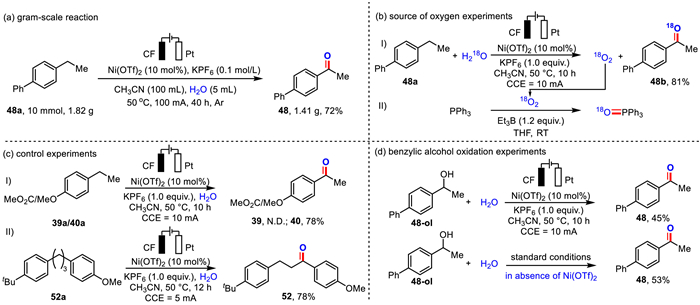
To gain insights into the mechanism of this transformation, we performed several mechanistic experiments. Firstly, we carried out the gram-scale reaction (Scheme 3a). As a result, the reaction of 4-ethyl-1,1′-biphenyl (10 mmol) under slightly modified conditions (40 h) gave 48 in 72% isolated yield (1.41 g). Then under standard conditions, the high yield of product 48b (81%) was obtained when H218O was used instead of ultrapure water (Scheme 3b, Ⅰ). The result indicated that the oxygen in the product comes from water. Subsequently, under the same conditions, H218O was also used for the energization reaction, and argon gas was continuously blown into the reaction and circulated through the system, resulting in triphenylphosphine oxide with 18O [75]. It was demonstrated that 18O2 could be generated in the standard system (Scheme 3b, Ⅱ). Next, a competition reaction was performed with the addition of methyl-4-ethylbenzoate (0.125 mmol) and 1-ethyl-4-methoxybenzene (0.125 mmol). The reaction provided 4-methoxyacetophenone in 78% yield, and it was observed that when there are two potential reaction sites on a molecule, the reaction occurs preferentially at the benzylic position of the methoxyl-substituted aryl, indicating that electron-rich aromatic rings were more susceptible to oxidation than electron-withdrawing ones (Scheme 3c), thus, the desired product was selectively obtained. Also, we found that the addition of nickel salts had little effect on the yield (53% vs. 45%) when 1-([1,1′-biphenyl]-4-yl)ethan-1-ol was used as the substrate under standard conditions, suggesting that the nickel salts might not be involved in the reaction of alcohols to ketones (Scheme 3d). In contrast, the significant decrease in the yield of 48 from 87% to 45% from the standard conditions shown in Scheme 3d, indicates that the main reaction path might not proceed via the alcohol intermediate. Furthermore, we investigated the proton/deuterium kinetic isotope effect of the reaction. Under competitive conditions, the value of kH/kD is 1.82; and in parallel conditions, the value of kH/kD is 1.67, which indicates that the C–H bond cleavage might be partially involved in the rate-determining step (Figs. S9 and S10 in Supporting information). A bipolar ultramicroelectrode (BUME) combined with nano-electrospray ionization mass spectrometry was used to rapidly capture short-lived reactive intermediates during the electrochemical reactions.

|
Download:
|
| Scheme 3. Mechanistic experiments: (a) Gram-scale reaction. (b) Source of oxygen experiments. (c) Control experiments. (d) Benzylic alcohol oxidation experiments. | |
{kind=link}
The fabrication of the bipolar ultramicroelectrode was developed in a previous study [76], and is only briefly introduced in Supporting information. A Cu wire (0.2 mm i.d.) was inserted into a quartz capillary from its rear end and then connected to a high-voltage power supply. The BUME took advantage of the high voltage to trigger electrospray ionization and to induce synergetic redox reaction at the two ends of the deposited carbon film inside of the spray tip (Fig. S11 in Supporting information). With this method, electrospray ionization and Faradaic reaction occurred simultaneously, permitting rapid transfer of the electrochemically generated short-lived intermediates into an LTQ-XL mass spectrometer (Thermo-Fisher, Waltham, MA) for analysis. If a positive high voltage is applied, oxidation half reactions occur on the far end of the carbon film that is closer to the sprayer tip, facilitating mass analysis. In this experiment, a positive voltage of 1024 V was applied to the Cu wire to induce redox reaction from a BUME nanopipette filled with 100 µmol/L 4-ethylanisole in acetonitrile. Only oxidation products can be observed with positive high voltage, as a result, the 4-ethylanisole radical cation (m/z 136) was immediately captured (Fig. S11), suggesting that this radical played a key role in initiating the reactions.
To gain further insights into the reaction mechanism, we performed preliminary mechanistic experiments utilizing voltammetry studies. Square wave voltammetry (SWV) studies were performed to investigate the electroreduction process of Ni(Ⅱ) to Ni(Ⅰ), and the results are depicted in Fig. S12a (Supporting information). An obvious peak around −1.78 V vs. Ag/Ag+ was observed (Fig. S12a, red line), which was consistent with a Ni(Ⅱ/Ⅰ) redox couple, indicating the Ni-circulation process in our reaction. We also conducted cyclic voltammetry (CV) studies to investigate the electro-oxidation process of a wide series of alkylarenes, and to observe how the position of oxidation peaks was closely related to the electronic effect of the substrates. For alkylarenes with electron-withdrawing groups on their benzene ring, such as methyl 4-ethylbenzoate, the low electron cloud density on the benzene ring caused this substrate to be harder to oxidize, and the corresponding peak (2.30 V vs. Ag/Ag+, Fig S12b in Supporting information, red line) was more positive than that of 4-ethylbiphenyl (1.95 V vs. Ag/Ag+, Fig. S12b, black line). While for electron-rich alkylarenes, such as 4-ethylanisole (1.49 V vs. Ag/Ag+, Fig. S12b, blue line), the corresponding oxidation peak was observed in the low potential range due to its high electro-oxidative reactivity.
Next, the electrocatalytic water-splitting performance of Ni(OTf)2 was characterized by linear sweep voltammetry (LSV) in a three-electrode cell, and the results of hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) were depicted in Figs. S12c and d (Supporting information), respectively. The polarization curves in Fig. S12c demonstrated that Ni(OTf)2 exhibited an OER operating overpotential (η20 at 20 mA/cm2, 1.35 V vs. Hg/HgO, performed in 1 mol/L KOH and 2.5 mmol/L Ni(OTf)2) lower than that of the Ni-free solution (1.58 V, performed in 1 mol/L KOH). The results indicated that Ni(OTf)2 could promote the oxygen evolution process, which might provide a higher concentration of oxygen source for product formation. A significant potential difference between the HER operating overpotential (η20 at 20 mA/cm2) of Ni(OTf)2 solution (−1.44 V vs. Hg/HgSO4, performed in 0.5 mol/L H2SO4 with 2.5 mmol/L Ni(OTf)2) and that of the Ni-free solution (−1.55 V, performed 0.5 mol/L H2SO4) was observed (Fig. S12d), indicating that Ni(OTf)2 could improve the cathode current efficiency, which promoted the anodic oxidation of the substrates simultaneously. Based on our experimental results and literature reports [72], a mechanism for paired electrochemical oxidation was proposed (Fig. 2).

|
Download:
|
| Fig. 2. Proposed main mechanistic pathways. | |
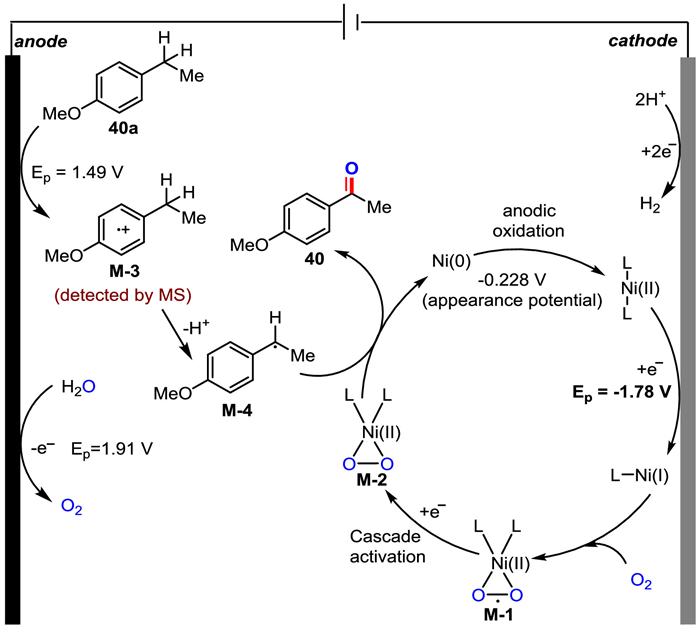{kind=link}
The anodic oxidation of substrate 40a produces radical cation M-3 (detected by MS, for details, see Supporting information) via single electron transfer (SET) and subsequent loss of a benzylic proton generates benzyl radical M-4. Concurrently, the Ni(Ⅰ) intermediate generated via one-electron reduction of the Ni(Ⅱ) species at the cathode activates O2 to afford Ni(Ⅱ)-superoxo species M-1. This Ni-superoxo species M-1 is then activated by a further one-electron reduction to provide the catalytically active Ni(Ⅱ)–peroxo species M-2, which could display oxygenase-like reactivity to transfer an oxygen atom to the external substrate M-4. The resulting reduced Ni(0) species undergo anodic oxidation to regenerate the Ni(Ⅱ) salt to complete the catalytic cycle and the final product 40 is formed. Yet other minor reaction pathways, such as M-4 reoxidized to benzyl cation which is then attacked by H2O, can at this point not be fully ruled out (path B, Fig. S13 in Supporting information).
In conclusion, we report the site-selective paired nickel-catalyzed electro-oxidation of alkyl aromatics by using water as the oxygen source to obtain valuable functionalized aryl carbonyl compounds and their derivatives with high selectivity and efficiency. This transformation proceeded smoothly under mild conditions with a wide range of substrates bearing various functional groups, such as free carboxylic acid, aldehyde, ester, halogen, alcohol, amide, and amino acid. It is also applicable to often sensitive and easily dehalogenated aryl iodides. Using green and clean water as the oxygen source provides a viable pathway for industrial scale-up production. In addition, further late derivatization of complex compounds and drug derivatives could open up opportunities for organic synthesis and medicinal chemistry. Mechanistic studies strongly supported the key role of the nickel catalyst in the selective C–H oxygenation, and the key aryl radical cation species involved in the reaction were also confirmed by a bipolar ultramicroelectrode (BUME) combined with nano-electrospray ionization mass spectrometry, which could be used to rapidly capture short-lived reactive intermediates. Further research on selective earth-abundant metal-catalyzed C–H oxidation is underway in the laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support from National Key R&D Program of China (No. 2022YFA1503200), National Natural Science Foundation of China (No. 22188101), the Fundamental Research Funds for the Central Universities (No. 63223007), Frontiers Science Center for New Organic Matter, Nankai University (No. 63181206) and Nankai University are gratefully acknowledged.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108660.
| [1] |
R. Francke, R.D. Little, Chem. Soc. Rev. 43 (2014) 2492-2521. DOI:10.1039/c3cs60464k |
| [2] |
Y. Jiang, K. Xu, C. Zeng, Chem. Rev. 118 (2018) 4485-4540. DOI:10.1021/acs.chemrev.7b00271 |
| [3] |
A. Jutand, Chem. Rev. 108 (2008) 2300-2347. DOI:10.1021/cr068072h |
| [4] |
R. Matthessen, J. Fransaer, K. Binnemans, et al., Beilstein J. Org. Chem. 10 (2014) 2484-2500. DOI:10.3762/bjoc.10.260 |
| [5] |
L.F.T. Novaes, J. Liu, Y. Shen, et al., Chem. Soc. Rev. 50 (2021) 7941-8002. DOI:10.1039/d1cs00223f |
| [6] |
J.L. Röckl, D. Pollok, R. Franke, et al., Acc. Chem. Res. 53 (2020) 45-61. DOI:10.1021/acs.accounts.9b00511 |
| [7] |
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230-13319. DOI:10.1021/acs.chemrev.7b00397 |
| [8] |
J.I. Yoshida, K. Kataoka, R. Horcajada, et al., Chem. Rev. 108 (2008) 2265-2299. DOI:10.1021/cr0680843 |
| [9] |
Y. Yuan, A. Lei, Acc. Chem. Res. 52 (2019) 3309-3324. DOI:10.1021/acs.accounts.9b00512 |
| [10] |
Q.L. Yang, P. Fang, T.S. Mei, Chin. J. Chem. 36 (2018) 338-352. DOI:10.1002/cjoc.201700740 |
| [11] |
N. Sauermann, T.H. Meyer, Y. Qiu, et al., ACS Catal. 8 (2018) 7086-7103. DOI:10.1021/acscatal.8b01682 |
| [12] |
C. Ma, P. Fang, Z.R. Liu, et al., Sci. Bull. 66 (2021) 2412-2429. DOI:10.1016/j.scib.2021.07.011 |
| [13] |
Y. Qiu, C. Zhu, M. Stangier, et al., CCS Chem. 3 (2021) 1529-1552. DOI:10.31635/ccschem.020.202000365 |
| [14] |
D.S. Chung, S.H. Park, S.G. Lee, et al., Chem. Sci. 12 (2021) 5892-5897. DOI:10.1039/d1sc00760b |
| [15] |
N. Fu, G.S. Sauer, A. Saha, et al., Science 357 (2017) 575-579. DOI:10.1126/science.aan6206 |
| [16] |
H. Huang, T.H. Lambert, J. Am. Chem. Soc. 143 (2021) 7247-7252. DOI:10.1021/jacs.1c01967 |
| [17] |
J. Ke, W. Liu, X. Zhu, et al., Angew. Chem. Int. Ed. 60 (2021) 8744-8749. DOI:10.1002/anie.202016620 |
| [18] |
Y. Qiu, M. Stangier, T.H. Meyer, et al., Angew. Chem. Int. Ed. 57 (2018) 14179-14183. DOI:10.1002/anie.201809611 |
| [19] |
T. Siu, C.J. Picard, A.K. Yudin, J. Org. Chem. 70 (2005) 932-937. DOI:10.1021/jo048591p |
| [20] |
F. Tang, K.D. Moeller, J. Am. Chem. Soc. 129 (2007) 12414-12415. DOI:10.1021/ja076172e |
| [21] |
H.C. Xu, K.D. Moeller, J. Am. Chem. Soc. 130 (2008) 13542-13543. DOI:10.1021/ja806259z |
| [22] |
Y. Yu, Y. Jiang, S. Wu, et al., Chin. Chem. Lett. 33 (2022) 2009-2014. DOI:10.1016/j.cclet.2021.10.016 |
| [23] |
L. Ackermann, Acc. Chem. Res. 53 (2020) 84-104. DOI:10.1021/acs.accounts.9b00510 |
| [24] |
K.J. Jiao, Y.K. Xing, Q.L. Yang, et al., Acc. Chem. Res. 53 (2020) 300-310. DOI:10.1021/acs.accounts.9b00603 |
| [25] |
F. Kakiuchi, T. Kochi, H. Mutsutani, et al., J. Am. Chem. Soc. 131 (2009) 11310-11311. DOI:10.1021/ja9049228 |
| [26] |
C. Ma, P. Fang, T.S. Mei, ACS Catal. 8 (2018) 7179-7189. DOI:10.1021/acscatal.8b01697 |
| [27] |
Y. Qiu, C. Tian, L. Massignan, et al., Angew. Chem. Int. Ed. 57 (2018) 5818-5822. DOI:10.1002/anie.201802748 |
| [28] |
A. Asghari, M. Ameri, A.A. Ziarati, et al., Chin. Chem. Lett. 26 (2015) 681-684. DOI:10.1016/j.cclet.2015.03.036 |
| [29] |
J.E. Bäckvall, A. Gogoll, J. Chem. Soc. Chem. Commun. (1987) 1236-1238. |
| [30] |
E.J. Horn, B.R. Rosen, Y. Chen, et al., Nature 533 (2016) 77-81. DOI:10.1038/nature17431 |
| [31] |
Y. Kawamata, M. Yan, Z. Liu, et al., J. Am. Chem. Soc. 139 (2017) 7448-7451. DOI:10.1021/jacs.7b03539 |
| [32] |
S.G. Robinson, J.B.C. Mack, S.N. Alektiar, et al., Org. Lett. 22 (2020) 7060-7063. DOI:10.1021/acs.orglett.0c01313 |
| [33] |
Z. Ruan, Z. Huang, Z. Xu, et al., Sci. China Chem. 64 (2021) 800-807. DOI:10.1007/s11426-020-9938-9 |
| [34] |
C. Feng, X. Liu, Y. She, et al., Chin. Chem. Lett. 34 (2023) 107935. DOI:10.1016/j.cclet.2022.107935 |
| [35] |
N.W.J. Ang, J.C.A. Oliveira, L. Ackermann, Angew. Chem. Int. Ed. 59 (2020) 12842-12847. DOI:10.1002/anie.202003218 |
| [36] |
M. Ishifune, H. Yamashita, M. Matsuda, et al., Electrochim. Acta 46 (2001) 3259-3264. DOI:10.1016/S0013-4686(01)00617-X |
| [37] |
Z. Li, W. Sun, X. Wang, et al., J. Am. Chem. Soc. 143 (2021) 3536-3543. DOI:10.1021/jacs.0c13093 |
| [38] |
L.L. Liao, Z.H. Wang, K.G. Cao, et al., J. Am. Chem. Soc. 144 (2022) 2062-2068. DOI:10.1021/jacs.1c12071 |
| [39] |
L. Ding, K. Niu, Y. Liu, et al., Chin. Chem. Lett. 33 (2022) 4057-4060. DOI:10.1016/j.cclet.2021.12.053 |
| [40] |
J. Xiang, M. Shang, Y. Kawamata, et al., Nature 573 (2019) 398-402. DOI:10.1038/s41586-019-1539-y |
| [41] |
P. Huang, P. Wang, S. Tang, et al., Angew. Chem. Int. Ed. 57 (2018) 8115-8119. DOI:10.1002/anie.201803464 |
| [42] |
L. Li, Y. Li, N. Fu, et al., Angew. Chem. Int. Ed. 59 (2020) 14347-14351. DOI:10.1002/anie.202006016 |
| [43] |
J.H. Qin, M.J. Luo, D.L. An, et al., Angew. Chem. Int. Ed. 60 (2021) 1861-1868. DOI:10.1002/anie.202011657 |
| [44] |
J.L. Röckl, D. Schollmeyer, R. Franke, et al., Angew. Chem. Int. Ed. 59 (2020) 315-319. DOI:10.1002/anie.201910077 |
| [45] |
H. Wang, X. Gao, Z. Lv, et al., Chem. Rev. 119 (2019) 6769-6787. DOI:10.1021/acs.chemrev.9b00045 |
| [46] |
Z.J. Wu, H.C. Xu, Angew. Chem. Int. Ed. 56 (2017) 4734-4738. DOI:10.1002/anie.201701329 |
| [47] |
J.J. Dai, X.X. Teng, W. Fang, et al., Chin. Chem. Lett. 33 (2022) 1555-1558. DOI:10.1016/j.cclet.2021.09.011 |
| [48] |
Z.W. Hou, M.M. Zhang, W.C. Yang, et al., J. Org. Chem. 87 (2022) 7806-7817. DOI:10.1021/acs.joc.2c00455 |
| [49] |
J. Kong, F. Zhang, C. Zhang, et al., Mol. Catal. 530 (2022) 112633. DOI:10.1016/j.mcat.2022.112633 |
| [50] |
X. Li, F. Bai, C. Liu, et al., Org. Lett. 23 (2021) 7445-7449. DOI:10.1021/acs.orglett.1c02651 |
| [51] |
L. Meng, J. Su, Z. Zha, et al., Chem. Eur. J. 19 (2013) 5542-5545. DOI:10.1002/chem.201204207 |
| [52] |
Y. Sun, X. Li, M. Yang, et al., Green Chem. 22 (2020) 7543-7551. DOI:10.1039/d0gc01871f |
| [53] |
P. Xiong, H.B. Zhao, X.T. Fan, et al., Nat. Commun. 11 (2020) 2706. DOI:10.1038/s41467-020-16519-8 |
| [54] |
C.C. Zeng, N.T. Zhang, C.M. Lam, et al., Org. Lett. 14 (2012) 1314-1317. DOI:10.1021/ol300195c |
| [55] |
M. Balaganesh, S. Lawrence, C. Christopher, et al., Electrochim. Acta 111 (2013) 384-389. DOI:10.1016/j.electacta.2013.08.020 |
| [56] |
Z. Li, Y. Zhang, K. Li, et al., Sci. China Chem. 64 (2021) 2134-2141. DOI:10.1007/s11426-021-1061-x |
| [57] |
J.A. Marko, A. Durgham, S.L. Bretz, et al., Chem. Commun. 55 (2019) 937-940. DOI:10.1039/c8cc08768g |
| [58] |
Y. Mo, K.F. Jensen, Chem. Eur. J. 24 (2018) 10260-10265. DOI:10.1002/chem.201802588 |
| [59] |
A.H. Cherney, N.T. Kadunce, S.E. Reisman, Chem. Rev. 115 (2015) 9587-9652. DOI:10.1021/acs.chemrev.5b00162 |
| [60] |
P. Gandeepan, T. Müller, D. Zell, et al., Chem. Rev. 119 (2019) 2192-2452. DOI:10.1021/acs.chemrev.8b00507 |
| [61] |
R. Jana, T.P. Pathak, M.S. Sigman, Chem. Rev. 111 (2011) 1417-1492. DOI:10.1021/cr100327p |
| [62] |
B. Liu, A.M. Romine, C.Z. Rubel, et al., Chem. Rev. 121 (2021) 14957-15074. DOI:10.1021/acs.chemrev.1c00519 |
| [63] |
S.W. Roh, K. Choi, C. Lee, Chem. Rev. 119 (2019) 4293-4356. DOI:10.1021/acs.chemrev.8b00568 |
| [64] |
B. Yang, Y. Qiu, J.E. Bäckvall, Acc. Chem. Res. 51 (2018) 1520-1531. DOI:10.1021/acs.accounts.8b00138 |
| [65] |
C. Amatore, C. Cammoun, A. Jutand, Synlett 2007 (2007) 2173-2178. DOI:10.1055/s-2007-985568 |
| [66] |
A. Das, J.E. Nutting, S.S. Stahl, Chem. Sci. 10 (2019) 7542-7548. DOI:10.1039/c9sc02609f |
| [67] |
T. Feng, S. Wang, Y. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202115178. DOI:10.1002/anie.202115178 |
| [68] |
P. Li, C. Guo, S. Wang, et al., Nat. Commun. 13 (2022) 3774. DOI:10.1038/s41467-022-31435-9 |
| [69] |
S. Wang, T. Feng, Y. Wang, et al., Chem. Asian J. 17 (2022) e202200543. DOI:10.1002/asia.202200543 |
| [70] |
Y. Wang, S. Tang, G. Yang, et al., Angew. Chem. Int. Ed. 61 (2022) e202207746. DOI:10.1002/anie.202207746 |
| [71] |
Y. Wang, Z. Zhao, D. Pan, et al., Angew. Chem. Int. Ed. 61 (2022) e202210201. DOI:10.1002/anie.202210201 |
| [72] |
Y. Liang, S.H. Shi, R. Jin, et al., Nat. Catal. 4 (2021) 116-123. DOI:10.1038/s41929-020-00559-w |
| [73] |
W.J. Kerr, A.J. Morrison, M. Pazicky, et al., Org. Lett. 14 (2012) 2250-2253. DOI:10.1021/ol300652k |
| [74] |
S.S. Weng, H.C. Li, T.M. Yang, RSC Adv. 3 (2013) 1976-1986. DOI:10.1039/C2RA23068B |
| [75] |
X. Liu, R. Liu, J. Qiu, et al., Angew. Chem. Int. Ed. 59 (2020) 13962-13967. DOI:10.1002/anie.202005765 |
| [76] |
J. Hu, N. Zhang, P.-K. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 18244-18248. DOI:10.1002/anie.202008577 |