 2023, Vol. 34
2023, Vol. 34 


b State Key Laboratory of New Textile Materials and Advanced Processing Technologies, Wuhan Textile University, Wuhan 430200, China
Advanced oxidation processes (AOPs) based on peroxymonosulfate (PMS) have attracted increasing attention over the past 10 years as an emerging approach for the degradation of persistent and refractory organic pollutants in water [1-3]. PMS is primarily activated in the radical pathway, where it produces sulfate radicals (SO4•−) and hydroxyl radicals (•OH), both of which are highly reactive and nonselective for most organic compounds [4,5]. The formation of additional reactive species, such as singlet oxygen (1O2) [6-8], high-valent metals [9-11], and metal–PMS complexes [12,13], can also activate PMS through non-radical pathways. These non-radical reactive species are more tolerant than radicals (SO4•− and •OH) and are more selective for organic molecules with high electron densities [14]. Owing to their special characteristics, PMS-based AOPs have greater flexibility in mitigating wastewater pollution by organic contaminants.
PMS self-activating radicals have low reactivities, meaning an effective activator is required to ensure PMS activation. In previous studies, Mn2+ was shown to effectively activate PMS to generate SO4•− and degrade pollutants, but it was difficult to achieve valence cycling, and the process could not sustainably degrade pollutants [15]. In view of this problem, manganese oxides have received considerable attention recently because of their environmental friendliness, cost-effectiveness, and excellent catalytic properties [16]. Mn2O3 is considered an efficient heterogeneous activator in the degradation of organic pollutants such as phenol [17] and tetrabromobisphenol [18]. In addition, Mn3O4 has been found to have greater catalytic activity and can activate PMS to completely remove ciprofloxacin [19]. The conventional hydrothermal method for preparing Mn3O4 requires a long reaction time, which varies from 48 h to 72 h at different temperatures and pressures [20]. The poor chemical and thermal stability of manganese oxides can also lead to nanoparticle (NP) aggregation, which reduces the catalytic efficiency [21].
To address these problems, Fan et al. [8] prepared manganese-doped graphite-phase carbon nitride (g-C3N4) materials and used them as catalysts to activate PMS for the degradation of acetaminophen, with a 100% removal efficiency within 15 min. Du et al. [22] used a manganese/magnetite/graphene oxide hybrid catalyst to activate PMS and produced SO4•− for the removal of bisphenol A (BPA). Derived carbon materials have various applications because of their distinct benefits, including low generation costs, spontaneously evolved structures, and flexible control over composition, structure, and morphology [23-25]. For instance, the pyrolysis of metal organic frameworks (MOFs) precursors can generate various novel carbonaceous materials with tunable contents, structures, and morphologies [26]. Liu et al. synthesized a core-shell MOF@COF combination on a titanium-based MOF to selectively functionalize carbonaceous materials [27]. Wang et al. [28] immobilized MIL-88A (Fe) particles on cotton fibers to improve the durability and stability of MIL-88A. Previous studies suggest the ability of composite catalysts to degrade antibiotics has also improved [29,30]. There has, to date, been limited research on activated carbon fiber (ACF) as carriers loaded and modulated with manganese oxides, intended to catalyze PMS for the degradation of organic contaminants, and to clarify this mechanism. ACF have long been utilized as electrodes for capacitors [31,32] and as adsorbents for wastewater treatment [33,34] because of their higher electrical conductivity in the gas or liquid phase and high adsorption rates. It has recently been found that ACF can also directly catalyze PMS to degrade pollutants [35,36].
In this study, we present a simple method for fabricating MnOx@ACF composites that can efficiently activate PMS for the removal of organic pollutants. Additionally, we conducted several experiments to demonstrate the role of MnOx@ACF as an adsorption catalyst for the degradation of organic pollutants. The development of aqueous and surface reactive species as well as their roles in pollutant degradation were investigated and assessed. Elements that might affect the emergence of active species and the degradation of pollutants were also examined.
The MnOx@ACF catalyst was synthesized using an in-situ oxidation anchoring method. ACF (0.5 g) was added to 200 mL of MnCl2·4H2O solution in a flask (500 mL). The suspension was stirred for 2 h to achieve the equilibrium uptake of Mn2+, that is, sufficient adsorption/ion exchange. A 100 mL mixture of KMnO4 and NaOH solution was slowly added to the suspension. After stirring for 4 h, the suspension was kept still and aged for 12 h for the complete in-situ formation and growth of MnOx onto the ACF. The catalyst was separated from the solution and washed several times with deionized water to remove excess KMnO4 and NaOH. The catalyst was then dried at 80 ℃ for 8 h in a drying oven. The product was finally calcined at 400 ℃ for 2 h in a muffle furnace and was subsequently referred to as MnOx@ACF in a later study. Detailed information on all chemicals, experimental procedures, material characterization methods, and analytical methods are provided in the Supporting information.
X-ray diffraction (XRD) measurements were performed to determine the phase structures of the MnOx@ACF and ACF. The fresh MnOx@ACF's XRD pattern is shown in Fig. S1a (Supporting information), where the peaks were attributed to the tetragonal Mn3O4's characteristic planes (JCPDS No. 80–0382). It is interesting to note that even when essential matches between the diffraction peaks and Mn3O4 were made, a new, weak diffraction peak at 12.5° was observed, which was linked to the presence of MnO2 (JCPDS No. 44–0141). This indicates that MnO2, an amorphous phase, did not fully crystallize and remained in the catalyst in an amorphous form, while Mn3O4 in the catalyst retained high phase purity [37].
The Fourier-transform infrared spectroscopy (FT-IR) spectra of the MnOx@ACF compounds are shown in Fig. S1b (Supporting information). Two main bands at approximately 626.5 and 504.9 cm−1 and the peaks typically assigned to ACF vanished in the spectrum of the fresh MnOx@ACF, suggesting the new bands were associated with the coupling modes between the Mn-O stretching modes of tetrahedral and octahedral sites [38]. All these results demonstrated the successful preparation of composite manganese-based nanomaterials [39].
The changes in surface morphology of Mn and ACF were observed using scanning electron micrographs and energy dispersive spectrometry elemental mapping analysis (Fig. 1). ACF was created by joining together several carbon fibers, as shown in Fig. 1a and Fig. S2 (Supporting information), and it featured a few distinct strip grooves on its surface. Owing to its unique structure, ACF has a substantial number of holes, which increases its surface area and provides additional necessary foam properties, including bendability and compressibility [40]. Minerals or salts in the biomass are sources of small, somewhat rough, bumps on the fiber surface [41]. In Fig. 1e, it can be seen that MnOx nanomaterials were successfully loaded into the strip grooves of the ACFs and that visible agglomerations developed on their surfaces. The results demonstrated that the nanoparticles may migrate from the ACF grooves to the outer surface of the carbon fibers, where the agglomeration becomes increasingly evident [42]. Energy dispersive spectrometry elemental mapping analysis was performed on the ACF and MnOx@ACF (Fig. 1), where the results showed that C, O, and Mn were uniformly distributed in the structure with atomic mass percentages of 94.71%, 5.28% and 0.01% of ACF and 76.38%, 20.52% and 3.11% of MnOx@ACF, respectively, demonstrating the successful formation of MnOx on top of the ACF.

|
Download:
|
| Fig. 1. Scanning electron micrographs and energy dispersive spectrometry elemental mapping of (a–d) activated carbon fiber (ACF) and (e–h) MnOx@ACF. | |
{kind=link}
The chemical composition of the prepared catalysts was determined using X-ray photoelectron spectroscopy (XPS), and the results are shown in Fig. S3 and Table S3 (Supporting information). In Fig. S3a, the Mn 2p and O 1s peaks appear broad because of the multiple spitting [43], whereas the presence of manganese oxide was confirmed by analyzing the XPS spectra of Mn 2p and O 1s of the catalyst. The spin-orbit splitting between Mn 2p3/2 (642.15 eV) and Mn 2p1/2 (653.56 eV) was 11.41 eV (Fig. S3b). The high-resolution Mn 2p spectrum was decomposed, and three peaks were clearly observed. The peaks at 641.26, 642.40, and 644.78 eV in the Mn 2p3/2 were assigned to the binding energies of Mn2+, Mn3+, and Mn4+, respectively. It could also be established that Mn2+, Mn3+, and Mn4+ accounted for 10.04%, 65.04%, and 24.92% of manganese species in MnOx@ACF, respectively. The molar ratio of Mn2+, Mn3+, and Mn4+ in the composite catalyst was therefore calculated to be 2:13:5, and the ratio of Mn3O4 to MnO2 was determined to be 1:1, consistent with a previous study [44].
As shown in Fig. S3c, the high-resolution O 1s spectrum could be divided into three peaks, corresponding to the various oxygen-containing chemical bonds [45,46]. The three peaks with binding energies at 530.16 eV, 531.25 eV, and 532.44 eV were indexed to the oxide (Mn−O−Mn), hydroxide (Mn−O−H), and carbon-oxygen (C=O), respectively [19,42,45]. The chemical valence of manganese oxide can also be calculated using Eq. 1. Peak areas of the Mn−O−Mn bond and Mn−OH bond are in the O 1s peak [42]. From Eq. 1, the valence of manganese oxide was calculated to be 3.1. This result is similar to the average valence (3.15) from the molar ratio of Mn ions analyzed by the Mn 2p spectrum.
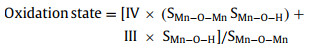
|
(1) |
The efficiency of TCH degradation was compared between the adsorption-catalytic activity of homogeneous and heterogeneous manganese species in various PMS systems, as indicated in Fig. S4a (Supporting information). In the PMS system without activation, PMS destroyed the electron-rich moieties of TCH by attacking the phenolic diketone and deprotonated dimethylamine groups to achieve partial degradation of TCH (21.7%) [47]. However, the other three systems, Mn2+/PMS, Mn3O4/PMS, and MnO2/PMS, achieved 75.5%, 64.8%, and 75.3% removal of TCH, respectively. Compared to that in the single PMS system, approximately 88.9% of the TCH was degraded within 60 min, and a significant amount (67.2%) was found in the MnOx@ACF/PMS system. Compared to other systems (Mn2+/PMS, Mn3O4/PMS, and MnO2/PMS), MnOx@ACF/PMS had a lower Mn input and better TCH degradation at the same catalyst dosage (0.1 g/L), indicating that MnOx@ACF/PMS had the potential for a low-cost and high-efficiency application. Table S4 (Supporting information) displays the TCs removal efficiencies of the various catalysts and systems.
To prove its long-term practical application, cyclical experiments were conducted (Fig. S4b in Supporting information). After five cycles, the degradation efficiency of TCH decreased by only approximately 16.2% (89.2%–73.0%). Moreover, the XRD results showed no significant change in the structure of MnOx@ACF before and after the reaction (Fig. S1a), indicating that the MnOx@ACF retains a stable phase structure. In the FT-IR spectra of the used MnOx@ACF, distinct diffraction peaks appeared at 1596 and 3430 cm−1. The former is attributed to the stretching vibration of the N−N bond in the amide group [48,49], whereas the latter is attributed to the C−O stretching mode in the ether and the O−H mode in the phenol group [35,50]. This might account for the degradation products produced by the ring-opening reaction absorbed on the surface of the catalyst [51]. Additionally, the MnOx@ACF after the reaction retained the fibrous structure, but the number of the surface particles were decreased which might be due to the adsorption of some intermediate TCH products on the surface of the ACF (Fig. S5 in Supporting information). The XPS spectra of the fresh and used MnOx@ACF were compared, and the results are shown in Fig. S3 and Table S3. The ion composition of MnOx@ACF did not change prior to and post use. From Eq. 1 and Table S3, the valence of the manganese oxides after the reaction was calculated to be 3.14, which was almost unchanged from that before the reaction, further demonstrating the stability and reusability of the MnOx@ACF.
To identify the aqueous reactive species generated in the MnOx@ACF/PMS system, electron paramagnetic resonance spectroscopy (EPR) analysis using 5, 5-dimethyl-1-pyrroline-N-oxide (DMPO) as a radical trapping agent for •OH and SO4•− was performed, and using methanol (MeOH) and tert-butanol (TBA) as radical scavengers to investigate the role of the reactive radicals in TCH degradation in quenching experiments. The reaction rate constants of the various radicals with the corresponding scavengers are listed in Table S5 (Supporting information). As can be seen in Fig. 2a and Fig. S6, there was no significant difference when varying the dosage of MeOH and TBA, and the degradation of TCH only decreased by 11.8% (88.9% to 77.1%) and 9.1% (88.9% to 79.8%) in the presence of 100 mmol/L MeOH and TBA, respectively. However, as shown in Fig. 2b, the strong EPR signal of DMPO−•OH (1:2:2:1 sextet) adducts, and weakened signal of the DMPO−SO4•− (1:1:1:1:1:1 sextet) adducts, can be clearly observed in the MnOx@ACF/PMS system. The results show that despite the fact that •OH and SO4•− were present in the system, neither played the major role in the degradation of TCH in the MnOx@ACF/PMS system.

|
Download:
|
| Fig. 2. (a) Effect of various quenching agents on tetracycline hydrochloride (TCH) degradation in the MnOx@ACF/PMS system. Electron paramagnetic resonance spectroscopy spectrum of (b) DMPO-SO4•−/•OH, (c) DMPO−O2•– and (d) TEMP-1O2 in the MnOx@ACF/peroxymonosulfate (PMS) system. (e) Degradation of different pollutants in the MnOx@ACF/PMS system and (f) transformation efficiency of PMS and removal efficiency of total oxidizing species in the PMS and MnOx@ACF/PMS systems. Initial conditions: [catalyst] = 0.1 g/L, [PMS] = 0.1 mmol/L, [DMPO] = [TEMP] = 100 mg/L, [TCH] = [Phenol] = [BPA] = [BA]= 20 mg/L and pH 5. | |
{kind=link}
Fig. 2c similarly shows the characteristic peaks of DMPO−O2•− in the MeOH solution. The strong signal peak of the adducts generated by 1O2 captured by 2, 2, 6, 6-tetramethyle-4-piperdone (TEMP) can also be detected in Fig. 2d. To further investigate whether 1O2 and O2•− determine TCH degradation, l-histidine and p-benzoquinone (BQ) were applied to the MnOx@ACF/PMS system. As shown in Fig. 2a, only 9.4% (60% to 50.6%) of TCH was removed after adsorption saturation in the presence of l-histidine. The formation of 1O2 could also be attributed to the reorganization of O2•−. To verify this, p-BQ was added to the reaction solution to allow observation of the effect of O2•− on the TCH degradation. With the addition of p-BQ, the degradation of TCH was almost completely inhibited, confirming the conversion of O2•− to 1O2 after generation. These results suggest that both 1O2 and O2•− dominated TCH degradation in the MnOx@ACF/PMS system.
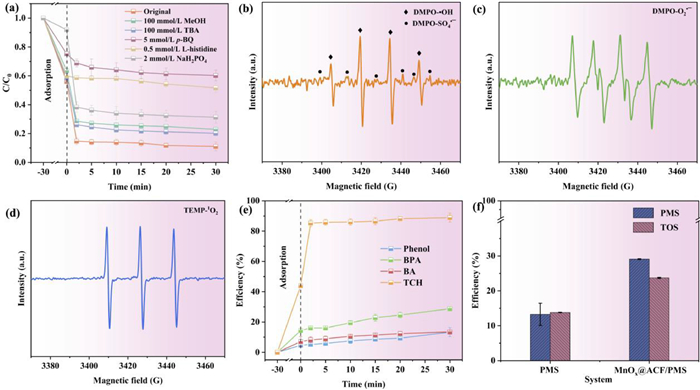
In previous reports, phosphate has been shown to have a strong affinity for surface −OH; phosphate can form an inner-sphere complex with transition metal ions to reduce the surface hydroxyl groups on the catalyst, thereby preventing interaction between the catalyst and PMS [52-54]. A phosphate-masking experiment was therefore conducted to investigate the role of surface hydroxyl groups in the catalyst, during degradation. As presented in Fig. 2a, 5 mmol/L NaH2PO4 significantly inhibited the efficiency of the MnOx@ACF/PMS system, and only approximately 21% (90.2% to 69.2%) of TCH was degraded, demonstrating that the surface −OH was essential for controlling the cleavage of peroxide bonds.
Surface reactive species, in addition to aqueous reactive species, may play an important role in contaminant degradation. The removal effectiveness of various organic contaminants in the MnOx@ACF/PMS system was variable, which can be seen in Fig. 2e. Approximately 88.9% of TCH and 28.8% of BPA were removed, whereas only 13.3% of phenol and BA were removed. Different active species can selectively oxidize certain structural compounds. For example, BPA has a low ionization potential and is easily degraded in both free radical and non-radical pathways [55]; BA is inert to non-radical oxidation and acts as a chemical probe for •OH and SO4•− [56], whereas phenol is more easily oxidized in the electron transfer state [57]. These results demonstrate that the MnOx@ACF/PMS system was highly selective for various contaminants and that •OH and SO4•− were not the dominant active species in the system [58]. We consequently developed the concept of total oxidizing species (TOS) [59], which encompasses both undecomposed PMS and other potential aqueous and surface reactive species generated by the transformation of PMS in the MnOx@ACF/PMS system. Detailed descriptions and experimental procedures of TOS and PMS are available in Text S3 (Supporting information). As shown in Fig. S7 (Supporting information), MnOx@ACF had almost no oxidation effect on KI. Approximately 29.1% of the PMS was transformed within 60 min in the MnOx@ACF/PMS system, while only 23.7% of the TOS was removed in the MnOx@ACF/PMS system, as shown in Fig. 2f. The obvious discrepancy between the TOS removal efficiency and PMS transformation efficiency indicated that a large proportion (23.7%/29.1% = 81.4%) of the decomposed PMS in the solution was transformed into short-lived aqueous reactive species (e.g., SO4•−, •OH, and 1O2), while a minor fraction (18.6%) decomposed into surface reactive species with relatively higher stability and accumulated in the system for a longer period. These observations led to the preliminary conclusion that the MnOx@ACF/PMS system generated both aqueous and surface reactive species.
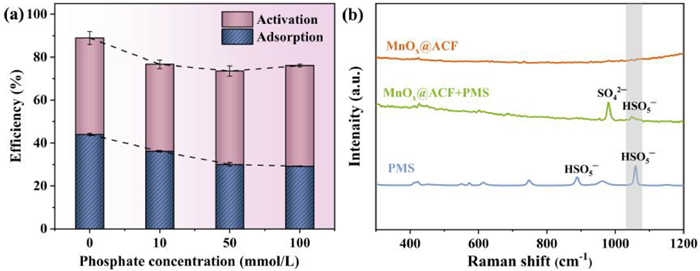
One possible surface reactive species is surface-complexed PMS. An increase in ionic strength decreases the zeta dot position of colloidal particles, which leads to the compression of the bilayer and affects electrostatic interactions (outer sphere interactions), while the effect on inner-sphere complexation is negligible [60-62]. Because of this, NaClO4 was used in this study to regulate the ionic strength, to evaluate the effect of the inner-sphere interactions on the degradation of TCH in the MnOx@ACF/PMS system. Fig. 3a indicates that the electrostatic adsorption of TCH by MnOx@ACF was inhibited when the concentration of NaClO4 was increased from 10 mmol/L to 100 mmol/L, although the activated degradation of TCH was almost unaffected by the addition of PMS. This suggests a strong inner-sphere interaction between PMS and the active sites on the catalyst surface.

|
Download:
|
| Fig. 3. (a) Effect of NaClO4 concentration on the degradation of tetracycline hydrochloride (TCH) and (b) Raman spectra of MnOx@ACF, MnOx@ACF+PMS, and peroxymonosulfate (PMS). Initial conditions: [catalyst] = 0.1 g/L, [PMS] = 0.1 mmol/L, [TCH] = 20 mg/L and pH 5. | |
{kind=link}
Raman spectroscopy was used to examine the interactions between the catalyst and PMS. Fig. 3b shows that MnOx@ACF did not exhibit a typical peak at a Raman shift of 300−1200 cm−1. The Raman spectrum of PMS showed two distinct peaks at 881 and 1060 cm−1, which might be attributed to HSO5− [62,63]. When MnOx@ACF was treated with the PMS solution, the S−O stretching in HSO5− changed from 1060 cm−1 to 1045 cm−1, indicating that the S−O stretching of PMS in interaction with the MnOx@ACF deteriorated the electron density of the S−O bond [64]. A fresh peak caused by SO42− appeared at 978 cm−1, indicating that MnOx@ACF and PMS interacted to form surface-complexed PMS.
An electrochemical analysis was conducted to further investigate the electron transfer process. To visually observe whether an electron transfer mechanism occurred in the MnOx@ACF/PMS system, a chrono-current analysis was performed. As shown in Fig. S8a (Supporting information), a slight current change was noted after the injection of PMS, that is, the active sites on the catalyst surface could effectively transfer electrons with PMS. However, it is worth pointing out that the change in current was not obvious after the addition of TCH. This implies that MnOx@ACF did not effectively mediate electron transfer from TCH to PMS; in other words, the electron transfer pathway was not the main route for the degradation of TCH in the MnOx@ACF/PMS system. Similarly, the linear sweep voltammetry curves in Fig. S8d (Supporting information) show that the simultaneous addition of PMS and TCH had only a slight effect on the current, which also confirmed the minimal electron transfer between the catalyst and PMS and TCH.
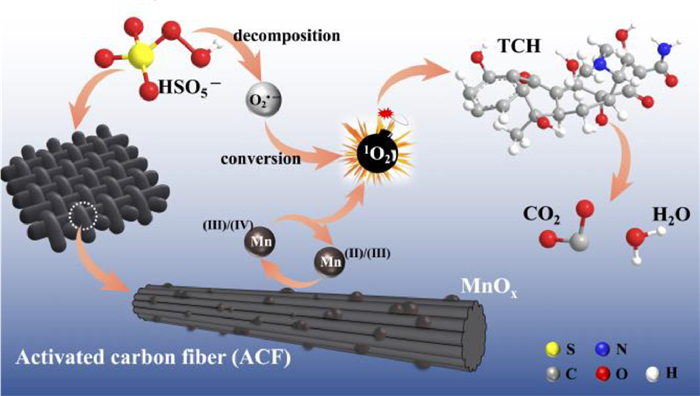
From this analysis, a possible mechanism of MnOx@ACF activation of PMS for TCH degradation was proposed. The detailed process of active species formation is summarized in the equations below and in Fig. 4. The enhanced activity of MnOx@ACF for PMS activation was attributed to the ideal coordinating sites for PMS, which were provided by the surface reactive Mn species of MnOx@ACF and the surface Mn(Ⅱ)/Mn(Ⅲ)/Mn(Ⅳ) cycle on the catalyst surface. PMS mainly exists in the form of HSO5− in solution [65] and provides negative charges. First, HSO5− dissociated from PMS could combine with surface reactive Mn species through OH−mediated interactions to form an inner-sphere complex (Eq. 2). After this, we noted the formation of O−O bridged species with the adjacent surface Mn(Ⅲ)/Mn(Ⅱ) under internal nucleophilic attack (Eq. 3), followed by the oxidative cleavage of O−O to produce two surface Mn(Ⅳ)/Mn(Ⅲ) species (Eq. 4). From this, we speculate that the formation of 1O2 may result from the reaction between ≡Mn(Ⅳ)/(Ⅲ)−OH and PMS. Analogous to Eqs. 2 and 3, the surface Mn(Ⅳ)/(Ⅲ) species provide sites for PMS to form an inner-sphere complex (≡Mn(Ⅳ)/(Ⅲ)−(O)OSO3−, Eq. 5). The complex can also trigger an internal nucleophilic attack to form an O−O bridge (Eq. 6). Electrons were subsequently transferred from the O−O bridge to the surface Mn(Ⅳ)/(Ⅲ) species, resulting in the release of 1O2 and regeneration of the surface Mn(Ⅲ)/Mn(Ⅱ) species (Eq. 7). Specifically, HSO5− might be directly adsorbed by amorphous MnO2 via ionization attraction and then generate SO4•− during the activation process (Eqs. 8 and 9) [66].
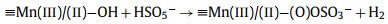
|
(2) |
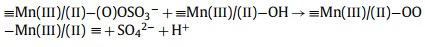
|
(3) |
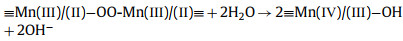
|
(4) |
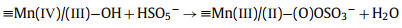
|
(5) |
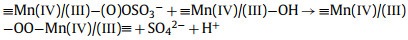
|
(6) |
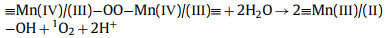
|
(7) |
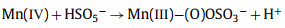
|
(8) |
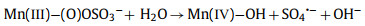
|
(9) |

|
Download:
|
| Fig. 4. Schematic illustration of MnOx@ACF activates peroxymonosulfate for tetracycline hydrochloride (TCH) degradation. | |
{kind=link}
During PMS activation and redox cycling of the surface Mn(Ⅱ)/Mn(Ⅲ)/Mn(Ⅳ), the generated reactive radicals attack the TCH molecules to decompose them. In addition, a portion of O2•− can be formed by the decomposition and conversion of PMS, which in turn generates 1O2 by recombination (see the details in Text S4 in Supporting information).
The mineralization of TCH during removal of the total organic carbon (TOC) in the MnOx@ACF/PMS system was explored. Fig. S10 (Supporting information) shows that after 60 min, approximately 35.1% of the TOC was removed from this system, indicating that mineralization was not complete and that additional small-molecule byproducts were present. The intermediates formed in the MnOx@ACF/PMS system during the degradation of TCH were identified using high performance liquid chromatography-mass spectrometry equipped with an electrospray pretexting (ESI) source, and the corresponding plots are shown in Fig. S11. From this, four possible pathways for explaining TCH degradation were proposed (Fig. S12 and Text S5 in Supporting information). Various influencing factors (including the MnOx loading content, doses of MnOx@ACF and PMS, contaminant concentration, pH of the solution, and presence of organic macromolecules and inorganic anions) were further evaluated, and the results and discussion are shown in Figs. S13, S14, Texts S6 and S7 (Supporting information).
In this work, a new efficient multivalent MnOx@ACF composite catalyst was prepared via a facile in-situ oxidation anchoring method, in which the partial conversion of MnO2 to Mn3O4 with ACF-doped MnOx@ACF had a large specific surface area, and rich adsorption and reaction sites were provided for Mn(Ⅱ)/Mn(Ⅲ) and Mn(Ⅲ)/Mn(Ⅳ), PMS, and TCH. On the ACF surface, cycling of Mn(Ⅱ)/Mn(Ⅲ) and Mn(Ⅲ)/Mn(Ⅳ) was encouraged, activating PMS to facilitate the degradation of TCH. In terms of which is the principal active species for TCH degradation in the MnOx@ACF/PMS system, 1O2 was the found to be the dominant active example, with a non-radical pathway contributing the majority of the oxidative component. Under conditions of pH 5, PMS dosing of 0.1 mmol/L, and MnOx@ACF dosing of 0.1 g/L, 88.9% TCH were removed. The degradation efficiency reached 73% after five cycles. In this study, we developed an Mn-based material capable of TCH degradation by activating PMS in a stable and effective manner. The oxidation process and degradation pathways were also fully investigated. MnOx is a common substance in the environment and is crucial for pollutant transformation. Future research should focus on manipulating MnOx@ACF using different surface engineering techniques, including oxygen vacancies, carbon defects, and crystallographic surfaces, with the aim of obtaining efficient and stable environmentally friendly catalysts.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this study.
AcknowledgmentThe authors thank the Technical Officer Zhengtao Gui of Shiyanjia Lab (www.shiyanjia.com) for the scanning electron microscopy and energy dispersive spectrometry elemental mapping analyses.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108407.
| [1] |
S. Xiao, M. Cheng, H. Zhong, et al., Chem. Eng. J. 384 (2020) 123265. DOI:10.1016/j.cej.2019.123265 |
| [2] |
M. Klavarioti, D. Mantzavinos, D. Kassinos, Environ. Int. 35 (2009) 402-417. DOI:10.1016/j.envint.2008.07.009 |
| [3] |
F. Ghanbari, M. Moradi, Chem. Eng. J. 310 (2017) 41-62. DOI:10.1016/j.cej.2016.10.064 |
| [4] |
Y. Zhang, J. Liu, A. Moores, et al., Environ. Sci. Technol. 54 (2020) 4631-4640. DOI:10.1021/acs.est.9b07113 |
| [5] |
C. Chu, J. Yang, X. Zhou, et al., Environ. Sci. Technol. 55 (2021) 1242-1250. DOI:10.1021/acs.est.0c06086 |
| [6] |
Y. Zhou, J. Jiang, Y. Gao, et al., Environ. Sci. Technol. 49 (2015) 12941-12950. DOI:10.1021/acs.est.5b03595 |
| [7] |
A. Jawad, K. Zhan, H. Wang, et al., Environ. Sci. Technol. 54 (2020) 2476-2488. DOI:10.1021/acs.est.9b04696 |
| [8] |
J. Fan, H. Qin, S. Jiang, Chem. Eng. J. 359 (2019) 723-732. DOI:10.1016/j.cej.2018.11.165 |
| [9] |
L. Wang, H. Xu, N. Jiang, et al., Environ. Sci. Technol. 54 (2020) 4686-4694. DOI:10.1021/acs.est.0c00284 |
| [10] |
H. Li, C. Shan, B. Pan, Environ. Sci. Technol. 52 (2018) 2197-2205. DOI:10.1021/acs.est.7b05563 |
| [11] |
H. Li, C. Shan, W. Li, et al., Water Res. 147 (2018) 233-241. DOI:10.1163/9781684170944_012 |
| [12] |
L. Wang, H. Xu, N. Jiang, et al., J. Hazard. Mater. 417 (2021) 126152. DOI:10.1016/j.jhazmat.2021.126152 |
| [13] |
H. Li, Z. Zhao, J. Qian, et al., Environ. Sci. Technol. 55 (2021) 6397-6406. DOI:10.1021/acs.est.1c02015 |
| [14] |
J. Lee, U. von Gunten, J.H. Kim, Environ. Sci. Technol. 54 (2020) 3064-3081. DOI:10.1021/acs.est.9b07082 |
| [15] |
G.P. Anipsitakis, D.D. Dionysiou, Environ. Sci. Technol. 38 (2004) 3705-3712. DOI:10.1021/es035121o |
| [16] |
J. Gu, P. Yin, Y. Chen, et al., Chin. Chem. Lett. 33 (2022) 4792-4797. DOI:10.1016/j.cclet.2022.01.029 |
| [17] |
E. Saputra, S. Muhammad, H. Sun, et al., Appl. Catal. B 142-143 (2013) 729-735. DOI:10.1016/j.apcatb.2013.06.004 |
| [18] |
S. Ndayiragije, Y. Zhang, Y. Zhou, et al., Appl. Catal. B 307 (2022) 121168. DOI:10.1016/j.apcatb.2022.121168 |
| [19] |
F. Wang, M. Xiao, X. Ma, et al., Chem. Eng. J. 404 (2021) 127097. DOI:10.1016/j.cej.2020.127097 |
| [20] |
S.K. Apte, S.D. Naik, R.S. Sonawane, et al., Mater. Res. Bull. 41 (2006) 647-654. DOI:10.1016/j.materresbull.2005.08.028 |
| [21] |
Y. Yao, C. Xu, S. Yu, et al., Ind. Eng. Chem. Res. 52 (2013) 3637-3645. DOI:10.1021/ie303220x |
| [22] |
J. Du, J. Bao, Y. Liu, et al., J. Hazard. Mater. 320 (2016) 150-159. DOI:10.1016/j.jhazmat.2016.08.021 |
| [23] |
T. Meng, Z. Li, Z. Wan, et al., Chem. Eng. J. 452 (2023) 139193. DOI:10.1016/j.cej.2022.139193 |
| [24] |
J. Guo, X. Xu, J.P. Hill, et al., Chem. Sci. 12 (2021) 10334-10340. DOI:10.1039/d1sc00915j |
| [25] |
T. Lu, Y. Liu, X. Xu, et al., Sep. Purif. Technol. 256 (2021) 117771. DOI:10.1016/j.seppur.2020.117771 |
| [26] |
Y. Zhang, J. Wu, S. Zhang, et al., Nano Energy 97 (2022) 107146. DOI:10.1016/j.nanoen.2022.107146 |
| [27] |
X. Liu, S. Zhang, G. Feng, et al., Chem. Mater. 33 (2021) 1657-1666. DOI:10.1021/acs.chemmater.0c04129 |
| [28] |
J.S. Wang, X.H. Yi, X. Xu, et al., Chem. Eng. J. 431 (2022) 133213. DOI:10.1016/j.cej.2021.133213 |
| [29] |
Z. Zhuge, X. Liu, T. Chen, et al., Chem. Eng. J. 421 (2021) 127838. DOI:10.1016/j.cej.2020.127838 |
| [30] |
J. Guo, L. Wang, X. Wei, et al., J. Hazard. Mater. 415 (2021) 125591. DOI:10.1016/j.jhazmat.2021.125591 |
| [31] |
H. Oda, A. Yamashita, S. Minoura, et al., J. Power Sources 158 (2006) 1510-1516. DOI:10.1016/j.jpowsour.2005.10.061 |
| [32] |
H. Li, J. Liang, H. Li, et al., J. Energy Chem. 31 (2019) 95-100. DOI:10.3390/rs12010095 |
| [33] |
S. Zhang, X.Y. Li, J.P. Chen, J. Colloid. Interface Sci. 343 (2010) 232-238. DOI:10.1016/j.jcis.2009.11.001 |
| [34] |
H. Tamai, T. Yoshida, M. Sasaki, et al., Carbon 37 (1999) 983-989. DOI:10.1016/S0008-6223(98)00294-2 |
| [35] |
S. Yang, L. Li, T. Xiao, et al., Appl. Surf. Sci. 383 (2016) 142-150. DOI:10.1016/j.apsusc.2016.04.163 |
| [36] |
S. Yang, T. Xiao, J. Zhang, et al., Sep. Purif. Technol. 143 (2015) 19-26. DOI:10.1016/j.seppur.2015.01.022 |
| [37] |
E. Saputra, S. Muhammad, H. Sun, et al., Catal. Commun. 26 (2012) 144-148. DOI:10.1016/j.catcom.2012.05.014 |
| [38] |
A.N. Chowdhury, M.S. Azam, M. Aktaruzzaman, et al., J. Hazard. Mater. 172 (2009) 1229-1235. DOI:10.1016/j.jhazmat.2009.07.129 |
| [39] |
J. Li, X. Li, X. Wang, et al., Appl. Catal. A: Gen. 584 (2019) 117170. DOI:10.1016/j.apcata.2019.117170 |
| [40] |
Q. Yang, L. Dong, C. Xu, et al., RSC Adv. 6 (2016) 12525-12529. DOI:10.1039/C5RA25701H |
| [41] |
S. Ye, G. Zeng, X. Tan, et al., Appl. Catal. B 269 (2020) 118850. DOI:10.1016/j.apcatb.2020.118850 |
| [42] |
W. Liu, P. Duan, X. Hu, et al., Ind. Eng. Chem. Res. 58 (2019) 22114-22123. DOI:10.1021/acs.iecr.9b04234 |
| [43] |
A. Moses Ezhil Raj, S.G. Victoria, V.B. Jothy, et al., Appl. Surf. Sci. 256 (2010) 2920-2926. DOI:10.1016/j.apsusc.2009.11.051 |
| [44] |
Z. Zhao, J. Zhao, C. Yang, Chem. Eng. J. 327 (2017) 481-489. DOI:10.1016/j.cej.2017.06.064 |
| [45] |
M. Chigane, M. Ishikawa, J. Electrochem. Soc. 147 (2000) 2246-2251. DOI:10.1149/1.1393515 |
| [46] |
L. Li, Q. Zhang, Y. She, et al., Sep. Purif. Technol. 270 (2021) 118770. DOI:10.1016/j.seppur.2021.118770 |
| [47] |
Y. Zhou, Y. Gao, J. Jiang, et al., Chem. Eng. J. 379 (2020) 122378. DOI:10.1016/j.cej.2019.122378 |
| [48] |
B. Zhang, L. Liu, L. Wang, et al., Carbon 134 (2018) 500-506. DOI:10.1016/j.carbon.2018.04.016 |
| [49] |
S. Li, S. Potana, D.J. Keith, et al., Chem. Commun. 50 (2014) 3931-3933. DOI:10.1039/c4cc00991f |
| [50] |
M.S. Shafeeyan, W.M.A.W. Daud, A. Houshmand, et al., J. Anal. Appl. Pyrol. 89 (2010) 143-151. DOI:10.1016/j.jaap.2010.07.006 |
| [51] |
Q. Wang, Z. Guan, S. Ding, et al., Sep. Purif. Technol. 289 (2022) 120625. DOI:10.1016/j.seppur.2022.120625 |
| [52] |
Y. Xu, J. Ai, H. Zhang, J. Hazard. Mater. 309 (2016) 87-96. DOI:10.1016/j.jhazmat.2016.01.023 |
| [53] |
T. Zhang, C. Li, J. Ma, et al., Appl. Catal. B: Environ. 82 (2008) 131-137. DOI:10.1016/j.apcatb.2008.01.008 |
| [54] |
A. Khan, H. Wang, Y. Liu, et al., J. Mater. Chem. A 6 (2018) 1590-1600. DOI:10.1039/C7TA07942G |
| [55] |
P. Duan, X. Liu, B. Liu, et al., Appl. Catal. B 298 (2021) 120532. DOI:10.1016/j.apcatb.2021.120532 |
| [56] |
P. Hu, H. Su, Z. Chen, et al., Environ. Sci. Technol. 51 (2017) 11288-11296. DOI:10.1021/acs.est.7b03014 |
| [57] |
W. Ren, L. Xiong, X. Yuan, et al., Environ. Sci. Technol. 53 (2019) 14595-14603. DOI:10.1021/acs.est.9b05475 |
| [58] |
Y. Yang, J. Jiang, X. Lu, et al., Environ. Sci. Technol. 49 (2015) 7330-7339. DOI:10.1021/es506362e |
| [59] |
H. Li, N. Yuan, J. Qian, et al., Environ. Sci. Technol. 56 (2022) 4498-4506. DOI:10.1021/acs.est.1c08790 |
| [60] |
Y. Xu, J. Ai, H. Zhang, J. Hazard. Mater. 309 (2016) 87-96. DOI:10.1016/j.jhazmat.2016.01.023 |
| [61] |
H. Lin, S. Li, B. Deng, et al., Chem. Eng. J. 364 (2019) 541-551. DOI:10.3390/md17100541 |
| [62] |
W. Tan, W. Ren, C. Wang, et al., Chem. Eng. J. 394 (2020) 124864. DOI:10.1016/j.cej.2020.124864 |
| [63] |
S. Shen, X. Zhou, Q. Zhao, et al., J. Hazard. Mater. 439 (2022) 129613. DOI:10.1016/j.jhazmat.2022.129613 |
| [64] |
L. Wang, H. Xu, N. Jiang, et al., J. Hazard Mater. 417 (2021) 126152. DOI:10.1016/j.jhazmat.2021.126152 |
| [65] |
Y.H. Guan, J. Ma, Y.M. Ren, et al., Water Res. 47 (2013) 5431-5438. DOI:10.1016/j.watres.2013.06.023 |
| [66] |
F. Pan, H. Ji, P. Du, et al., J. Hazard. Mater. 402 (2021) 123779. DOI:10.1016/j.jhazmat.2020.123779 |