 2023, Vol. 34
2023, Vol. 34 


b Department of Medicinal Chemistry, School of Pharmacy, Second Military Medical University, Shanghai 200433, China;
c State Key Lab of Anti-Infectives, Shanghai Institute of Pharmaceutical Industry, China State Institute of Pharmaceutical Industry, Shanghai 201203, China;
d IBMM, Faculty of Pharmacy, Montpellier University, CNRS, ENSCM, 34093 Montpellier Cedex 5, France
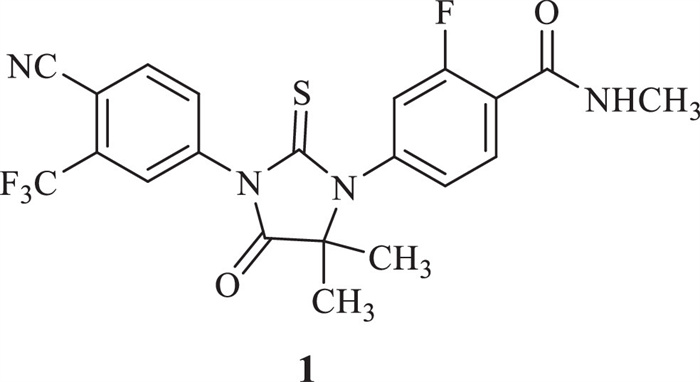
According to the statistics of the American Cancer Society 2022 [1], the number of prostate cancer (PCa) new cases is estimated at 268, 490 in US for 2022. Enzalutamide (1, Fig. 1) was the first second-generation non-steroidal androgen receptor (AR) antagonist to be used to treat castration-resistant prostate cancer (CRPC) that had spread or relapsed. Comparing with the first-generation AR antagonists nilutamide [2], flutamide [3] and bicalutamide [4], enzalutamide not only competitively inhibited androgen binding to AR, but also inhibited nuclear translocation of the AR, DNA binding and coactivator recruitment [5,6]. Enzalutamide was approved by FDA in 2012. Furthermore, enzalutamide showed potential therapeutic effect on triple-negative breast cancer with AR high-expression [7-10]. In the future, the requirement of active pharmaceutic ingredient (API) would be further increased. Therefore, it was of great research significance to develop a novel synthetic method of enzalutamide, especially that suitable to industrial production.

|
Download:
|
| Fig. 1. Structure of enzalutamide (1). | |
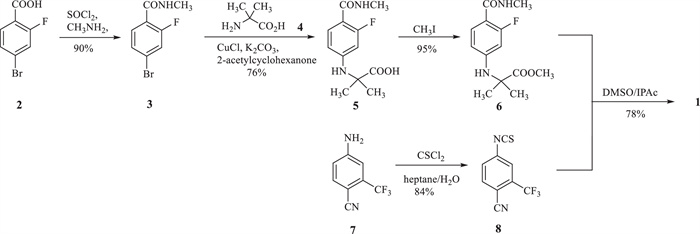
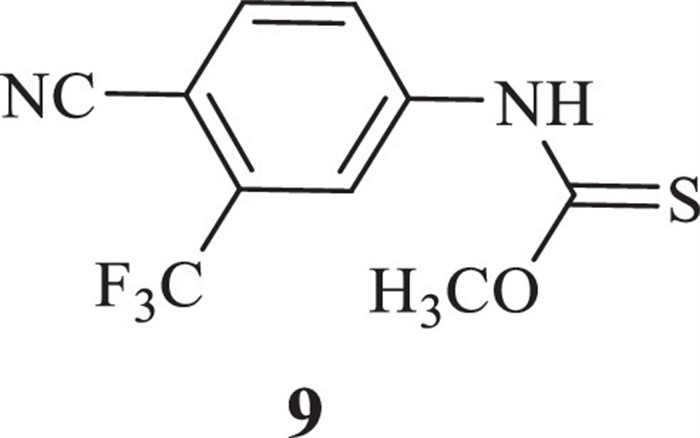
Two feasible routes have been reported for the synthesis of industrial scale enzalutamide. One was undertaken by researchers of Medivation Inc. (Scheme 1) [11], which began with 4-bromo-2-fluorobenzonic acid (2). The key intermediate methyl 2-((3-fluoro-4-(methylcarbamoyl)phenyl)-amino)-2-methylpropanoate (6) was prepared by chloration, amination, coupling reaction and methylation from starting material 2. Intermediate 6 reacted with 4-isothiocyanato-2-(trifluoromethyl)benzonitrile (8), which was made from 4-amino-2-(trifluoromethyl)benzonitrile (7), to obtain target compound 1. An evident disadvantage of this route was the use of the poisonous reagent iodomethane to prepare the intermediate 6. Zhou et al. [12] made an improvement for this route, but it was difficult to decrease the equivalent of isothiocyanate intermediate 8. Depending to the high reactivity of isothiocyanate intermediate 8, leaving group methanol, as a strong nucleophile byproduct of cyclization, could immediately react with 8 to form a typical impurity 9 (Fig. 2). Although impurity 9 could be easily removed by recrystallization with isopropanol, 2 equiv. of 8 were consumed for the cyclization step, at least.

|
Download:
|
| Scheme 1. The original route of Medivation Inc. for the synthesis of enzalutamide 1. | |

|
Download:
|
| Fig. 2. Typical impurity 9, by-product of Scheme 1 in cyclization. | |
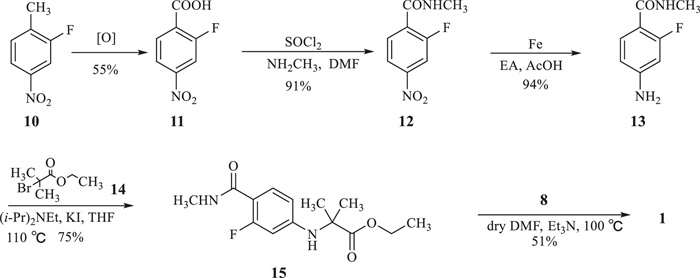
An alternative route was reported (Scheme 2) [13,14] to begin with 2-fluoro-4-nitrotoluene (10), following by oxidation, chloration, reduction, substitution and cyclization to afford target compound 1. Although toxic iodomethane was avoided in this route, starting material 10 and intermediates (11, 12) with a nitro group increased the risk of explosion during storage and transportation. Meanwhile, the reduction of 12 with Fe/AcOH or Pb/C-H2 was a high-risk reaction to synthesize intermediate 13 for industrial scale manufacture. Although API 1 could be obtained by the above routes, several environmentally unfriendly reagents, high-risk reactions and reactive intermediates were still utilized in these routes. Various synthetic strategies have been designed to overcome such disadvantages [15,16], however the low yield and high cost of the overall process limited the practical use of these routes. Thus, a novel synthetic route is urgently needed to develop for the efficient synthesis of 1 with mild condition, stable intermediate and environmentally friendly reagent. Herein we report a new route that highly limits toxic reagents and inconvenient reactive intermediates.

|
Download:
|
| Scheme 2. Alternative route for API 1 manufacture. | |
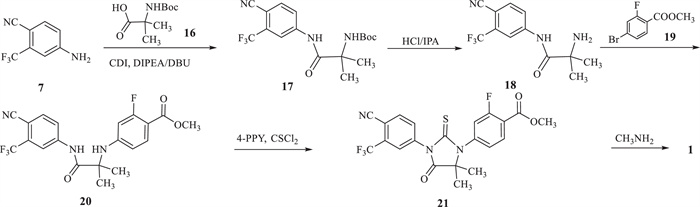
Our new route (Scheme 3) [17] begins with intermediate 7 and Boc-2-aminoisobutyric acid (16) to obtain tert-butyl (1-((4-cyano-3-(trifluoromethyl)phenyl)amino)-2-methyl-1-oxopropan-2-yl)carbamate (17) by condensation. The deprotection of 17 is carried out in the acidic condition to afford 2-amino-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-methylpropanamide (18). The third step is an Ullmann reaction for the coupling of intermediate 18 with methyl 2-fluoro-4-bromobenzoate (19) to give methyl 4-((1-((4-cyano-3-(trifluoromethyl)phenyl)amino)-2-methyl-1-oxopropan-2-yl)amino)-2-fluorobenzoate (20). Intermediate 20 is reacted with thiophosgene to obtain methyl 4-(3-(4-cyano-3-(trifluoromethyl)phenyl)-5,5-dimethyl-4-oxo-2-thioxoimidazolidin-1-yl)-2-fluorobenzoate (21). Enzalutamide (1) is finally prepared by methylamination of compound 21.

|
Download:
|
| Scheme 3. A novel route to synthesize enzalutamide. | |
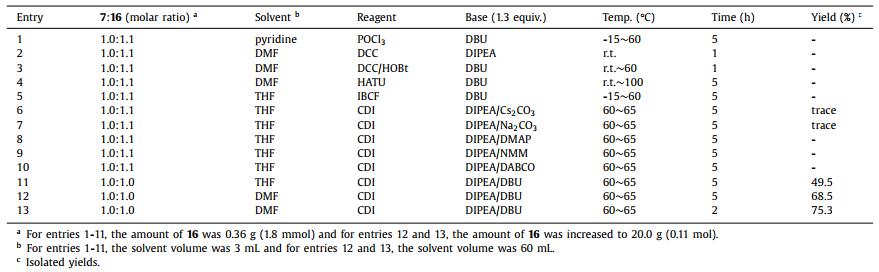
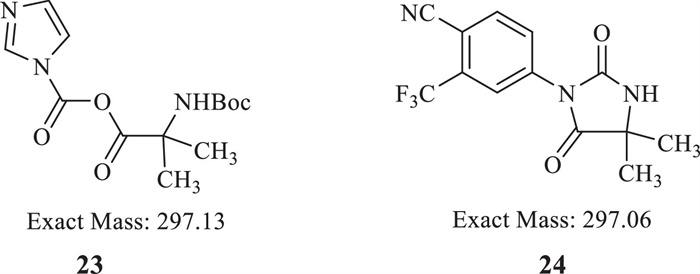
Because of the weak nucleophilicity of 7 with two strong withdrawing groups, several conditions were screened with phosphorus oxychloride (POCl3) [18], dicyclohexylcarbodiimide (DCC) [19-22], O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (HATU) [23] and isobutyl chloroformate (IBCF) [24]. Unfortunately, no target compound was found (Table 1, entries 1-5). When 1,1′-carbonyldiimidazole (CDI) was used to activate the carboxy group of starting material 16, trace amounts of intermediate 17 were found in entries 6 and 7. Because of the poor solubility of Cs2CO3 and Na2CO3 in THF, we tried to use organic alkali to make a homogeneous reaction such as 4-dimethylaminopyridine (DMAP), 4-methylmorpholine (NMM) and triethylenediamine (DABCO) (entries 8-10). Because of the weak alkaline, we failed to gain the intermediate 17. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was successfully used to obtain compound 17 in moderate yield by crystallization (entry 11). A typical impurity was found and isolated in the condensation step, with a mass signal [M + H]+ of 298.0. According to the mass result, two possible structures were speculated as reactive intermediate 23 or intramolecular closed ring product 24 (Fig. 3). 1H NMR, 13C NMR and HRMS were used to further identify the structure of this typical impurity (Supporting information). 1H NMR showed signals of aromatic hydrogens and no signal of the Boc protecting group. Therefore, we speculated that the structure of typical impurity was compound 24. In order to decrease the loss in mother liquid, N,N-dimethylformamide (DMF) was used instead of tetrahydrofuran (THF) (entry 12). We also found the typical impurity was increasing with the prolonged reaction time. Then the reaction time was shorted from 5 h to 2 h to control the amount of typical impurity (entry 13) and isolate yield was around 75.3%.
|
|
Table 1 Screening of conditions of the condensation step. |

|
Download:
|
| Fig. 3. Speculated structures of typical impurity 23 or 24. | |
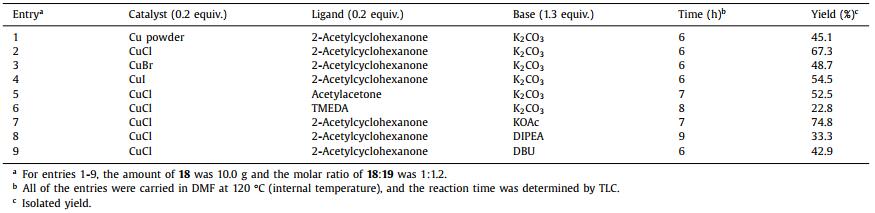
The deprotection of intermediate 17 was carried out with 4 mol/L HCl in isopropyl alcohol (IPA) [25] or trifluoroacetic acid (TFA) [26] to afford deprotected product 18 with 98.0% or 97.3% yield, respectively. Intermediate 18 was reacted with 19 by Ullmann coupling. Considering the cost of this route, copper sources were utilized as the catalyst in the coupling step [27]. Primarily, copper sources were screened such as copper powder, CuCl, CuBr and CuI (Table 2, entries 1-4) with 2-acetylcyclohexanone and K2CO3 in DMF under 120 ℃. Entry 2 showed a better yield at 67.3%. Secondly, we investigated ligands 2-acetylcyclohexanone (entry 2), acetylacetone and N,N,N',N'-tetramethylethylenediamine (TMEDA) (entries 5 and 6), in which 2-acetylcyclohexanone was still a suitable ligand in the coupling reaction. When this reaction was monitored by thin-layer chromatography (TLC), starting material 7 was found in this system. Therefore, we speculated that the strong basicity or poor stirring condition of K2CO3 make compound 18 or 20 hydrolyze. Finally, we screened bases KOAc, N,N-diisopropylethylamine (DIPEA) and DBU. The stirring conditions were obviously improved, when organic alkali was used (entries 8 and 9), product 20 was obtained in moderate yields (33.3% and 42.9%). The yield was increased to 74.8% when KOAc was used instead of K2CO3 (entry 7).
|
|
Table 2 Optimization of Ullmann coupling reaction. |
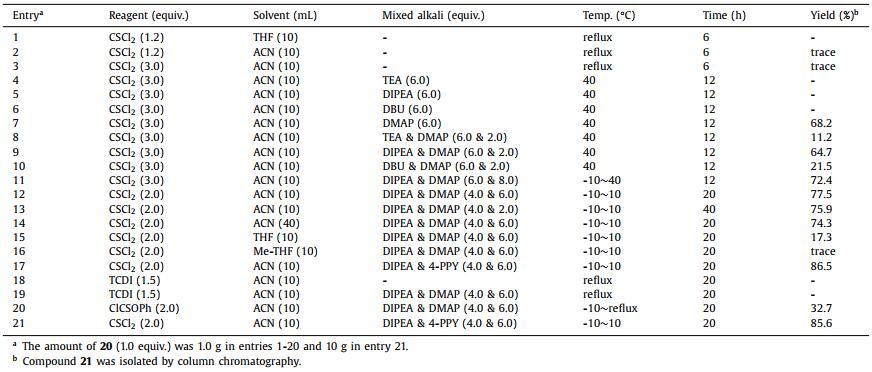
Coupling product 20 was reacted with thiophosgene (CSCl2) or thiophosgene derivatives to construct the thiohydantoin ring. The reaction conditions for cyclization were optimized by the following steps (Table 3). The initial conditions of 1.2 equiv. of thiophosgene and compound 20 in THF or acetonitrile (ACN) (entries 1 and 2) [11] gave only traces of the desired product in entry 2. Increasing the amount of thiophosgene (entry 3) to 3.0 equiv. failed to improve the conversion of intermediate 20. Because of the formation of byproduct HCl, we screened acid acceptors triethylamine (TEA), DIPEA and DBU (entries 4-6). However, no desired product was found. Notably, when DMAP was used as an acid acceptor, we successfully obtained the cyclization product 21 in 68.2% yield (entry 7). This result showed that DMAP was probably not only used as an acid acceptor, but also as a catalyst for cyclization. Byproduct HCl reacted with DMAP to form a quaternary ammonium salt, which had poor solubility in the solvent and made the stirring condition worse. Mixed alkali including DMAP were screened (entries 8-10) and a 64.7% yield was found with the mixed alkali DIPEA/DMAP. Increasing the proportion of DMAP to 8.0 equiv. improved the yield to 72.4% (entry 11). To further improve stirring conditions, equivalents of thiophosgene and DMAP were decreased to obtain target compound 21 in yield 75.9%-77.5% at lower reaction temperature in order to avoid thiophosgene decomposition (entries 12 and 13). Because cyclization was an intramolecular nucleophilic reaction, the volume of solvent ACN was increased to 40 mL (entry 14). The stirring condition was improved to obtain a 74.3% yield. Non-protonic solvents such as THF and 2-methyltetrahydrofuran (Me-THF) were screened but gave poor yields (entries 15 and 16). 4-Pyrrolidinopyridine (4-PPY), which is a DMAP analogue, was used as an alternative base in cyclization to give a better yield 86.5% (entry 17). 1,1′-Thiocarbonyldiimidazole (TCDI) [28] and phenyl chlorothionocarbonate (ClCSOPh) [29] were investigated to replace the thiophosgene (entries 18-20), which is an environment-unfriendly reagent. The reaction failed with TCDI (entries 18 and 19) and gave only 32.7% of desired product with ClCSOPh (entry 20). When ClCSOPh was used, diphenylthionocarbonate was found as a main byproduct. One molecule of phenol was released by this process, which was still a strong nucleophile and then reacted with another molecule of ClCSOPh to give byproduct diphenylthionocarbonate. This reaction required at less 2 equiv. of ClCSOPh and the byproduct was hard to remove from the crude product. Finally, we chose the typical condition to scale up to 10 g and obtain the desired product in 85.6% yield (entry 21). The last step was an amination, for which we referred to the method reported by Zhou et al. [12] to obtain enzalutamide in 86.8% yield.
|
|
Table 3 Optimization of the cyclization step. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion, a novel route to synthesize enzalutamide was established in five steps including condensation, deprotection, Ullmann coupling, cyclization and amination. Compared with the reported methods, unstable intermediate, toxic chemical and high-risk reaction were avoided in this new route. The purification of intermediate 21 needs to be further optimized. In the future, the challenge in manufacturing enzalutamide would be to completely abandon the use of thiophosgene. Taken together, this approach is a more efficient, convenient and economical route, which provides a new strategy for improving the process of API enzalutamide manufacture.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the Academic Mentorship for. This work was supported by Scientific Research Cadre Project (AMSCP) 2021 and National Key Research and Development Program of China (No. 2020YFA0509200 to C. Sheng).
| [1] |
R.L. Siegel, K.D. Miller, H.E. Fuchs, et al., CA Cancer J. Clin. 72 (2022) 7-33. DOI:10.3322/caac.21708 |
| [2] |
A. Decensi, D. Guameri, M.C. Padetti, et al., Eur. J. Cancer 27 (1991) 1100-1104. DOI:10.1016/0277-5379(91)90301-S |
| [3] |
E.J. Small, N.J. Vogelzang, J. Clin. Oncol. 15 (1997) 382-388. DOI:10.1200/JCO.1997.15.1.382 |
| [4] |
C.J. Tyrrel, A.V. Kaisary, P. Iversen, et al., Eur. Urol. 33 (1998) 447-456. DOI:10.1159/000019634 |
| [5] |
A. Rodriguez-Vida, M. Galazi, S. Rudman, et al., Drug Des. Dev. Ther. 9 (2015) 3325-3339. DOI:10.2147/DDDT.S69433 |
| [6] |
Y.S. Ha, S. Goodin, R.S. Dipaola, et al., Drugs Today 49 (2013) 7-13. DOI:10.1358/dot.2013.49.1.1910724 |
| [7] |
T.A. Traina, K. Miller, D.A. Yardley, et al., J. Clin. Oncol. 36 (2018) 884-890. DOI:10.1200/jco.2016.71.3495 |
| [8] |
A.Murray F.Caiazza, S.F. Madden, et al., Endocr. Relat. Cancer 23 (2016) 323-334. DOI:10.1530/ERC-16-0068 |
| [9] |
V.N. Barton, N.C. D'Amato, M.A. Gordon, et al., Mol. Cancer Ther. 14 (2015) 769-778. DOI:10.1158/1535-7163.MCT-14-0926 |
| [10] |
A. Anestis, P. Sarantis, S. Theocharis, et al., J. Cancer Res. Clin. Oncol. 145 (2019) 1221-1233. DOI:10.1007/s00432-019-02872-9 |
| [11] |
R.P. Jain, R. Angelaud, A. Thompson, et al., PCT Int. Appl. WO 2011106570, 2011.
|
| [12] |
A.N. Zhou, B.N. Li, L.J. Ruan, et al., Chin. Chem. Lett. 28 (2017) 426-430. DOI:10.1016/j.cclet.2016.09.007 |
| [13] |
R.S. Rathore, V.V. Durvasula, A. Sharma, et al., PCT Int. Appl. WO 2015063720 (2015).
|
| [14] |
L.J. Song, Y. Wang, X.F. Lu, et al., Fine Chem. Intermed. 42 (2012) 34-36. |
| [15] |
M.E. Jung, S Ouk, D. Yoo, et al., J. Med. Chem. 53 (2010) 2779-2796. DOI:10.1021/jm901488g |
| [16] |
P.H. Desai, S. Seetharaman, V.H. Nikam, et al., PCT Int. Appl. WO 2019106691, 2019.
|
| [17] |
X.G. Meng, L. Shao, G. Huang, et al., CN 109651256, 2019.
|
| [18] |
D.T.S. Rijkers, H.P.H.M. Adams, H.C. Hemker, et al., Tetrahedron 51 (1995) 11235-11250. DOI:10.1016/0040-4020(95)00671-T |
| [19] |
R.W. Driver, T.D.W. Claridge, S. Scheiner, et al., Chem. Eur. J. 22 (2016) 16513-16521. DOI:10.1002/chem.201602905 |
| [20] |
R.Z. Huang, B. Zhang, X.C. Huang, et al., RSC Adv. 7 (2017) 8866-8878. DOI:10.1039/C6RA25590F |
| [21] |
A. Jeganathan, S.K. Richardson, R.S. Mani, et al., J. Org. Chem. 56 (1986) 5362-5367. DOI:10.1021/jo00376a057 |
| [22] |
W. Wang, W.F. Li, Y.Q. Yang, et al., Huaxue Shiji 30 (2008) 185-190. |
| [23] |
X.B. Bi, J. Yin, C. Rao, et al., Org. Lett. 20 (2018) 7790-7793. DOI:10.1021/acs.orglett.8b03195 |
| [24] |
P. Gisbert, P. Trillo, I.M. Pastor, ChemistrySelect 3 (2018) 887-893. DOI:10.1002/slct.201702818 |
| [25] |
C. Chen, X.Y. Yang, H. Fang, et al., Eur. J. Med. Chem. 181 (2019) 111563. DOI:10.1016/j.ejmech.2019.111563 |
| [26] |
F. Wen, Z. Li, Synth. Commun. 50 (2020) 3462-3474. DOI:10.1080/00397911.2020.1802759 |
| [27] |
Z.K. Lu, R.J. Twieg, Tetrahedron Lett. 46 (2005) 2997-3001. DOI:10.1016/j.tetlet.2005.03.027 |
| [28] |
S.F. Zhang, X.H. Hou, L.L. Wang, et al., CN 114014850A, 2022.
|
| [29] |
C.B. Haim, A. Horvath, J.E.E. Weerts, PCT Int. Appl. WO 2016100652A2 2016.
|