 2023, Vol. 34
2023, Vol. 34 


b University of Chinese Academy of Sciences, Beijing 100049, China
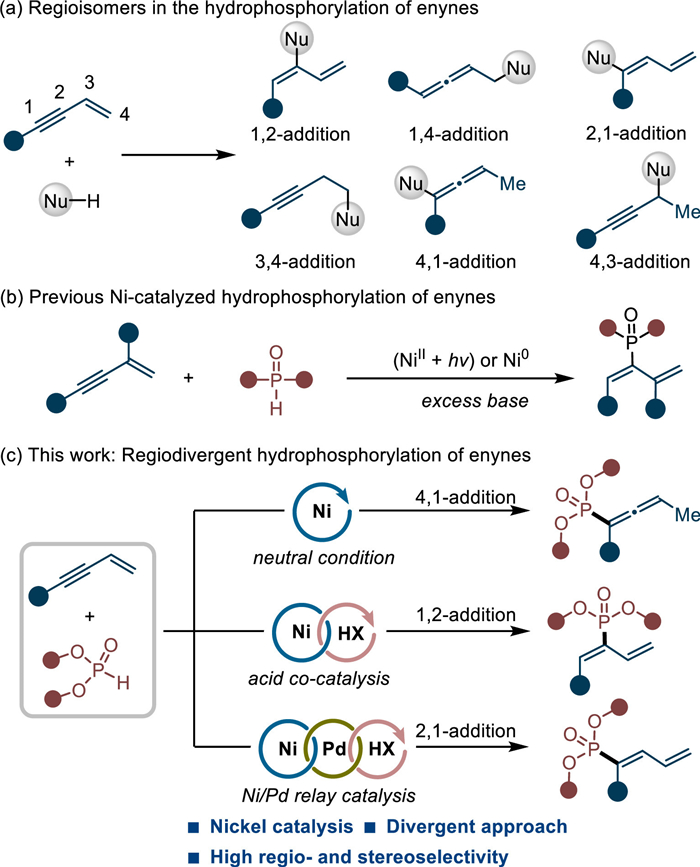
Owing to the versatile biological activities, organophosphorus has wide applications in medicinal and agricultural chemistry [1–5]. Besides, many phosphine compounds also serve as useful building blocks, ligands as well as organocatalysts in numerous synthetic transformations [6–9]. Therefore, considerable efforts have been devoted to their selective synthesis under catalysis [10–25]. Among all the developed protocols, the transition-metal catalyzed hydrophosphorylation of unsaturated C–C bonds represents one of the most atom-economic and straightforward tools [26–31]. In this regards, many intellectual achievements have been contributed to the selective hydrophosphorylation of alkenes [32–38], alkynes [39–53], allenes [54–56] and 1,3-dienes [57–61] over the past years. However, the regioselective hydrophosphorylation of conjugated enynes was rather rare [62,63]. Compared with alkene or alkyne surrogates, the hydrophosphorylated products from enynes usually contain at least two unsaturated C–C bonds that can facilitate the rapid assembly of molecular complexity by their further modification.
The regiodivergent hydrofunctionalization of enynes is by no means an easy work and considerable hurdles impede its development [64–67]. The competitive insertion between alkene and alkyne bonds often complicates most of the established methods. The existence of various π-metal tautomerisms also poses substantial challenges in regioselective control (Scheme 1a). Although some success have been achieved to forge the C–C [68–76], C–N [77–79], C–Si [80,81] or C–B [82–86] bonds, the selective construction of C–P bonds is limited. Recently, Zhu and co-workers developed a light-induced nickel catalysis for the 1,2-hydrophosphorylation of conjugated enynes with phosphine oxides in the presence of excess base [62]. Very recently, Zhang's group also disclosed a base-required nickel catalysis for 1,2-hydrophosphorylation of conjugated enynes (Scheme 1b) [63]. It should be noted that only one major isomer in hydrophosphorylation could be accessed under the above two protocols. On the basis of our continuous research interests in functionalization of alkenes/alkynes [87–92], herein we reported a regiodivergent strategy for selective hydrophosphorylation of conjugated enynes with phosphites (Scheme 1c). By manipulating the order of elementary reaction step, switchable regulation of 4,1- or 1,2-addition could be realized, respectively. With the help of an additional Pd–H catalysis, 2,1-hydrophosphorylation was also an accessible task in our protocol.

|
Download:
|
| Scheme 1. Catalytic regioselective hydrophosphorylation of enynes. | |
{kind=link}
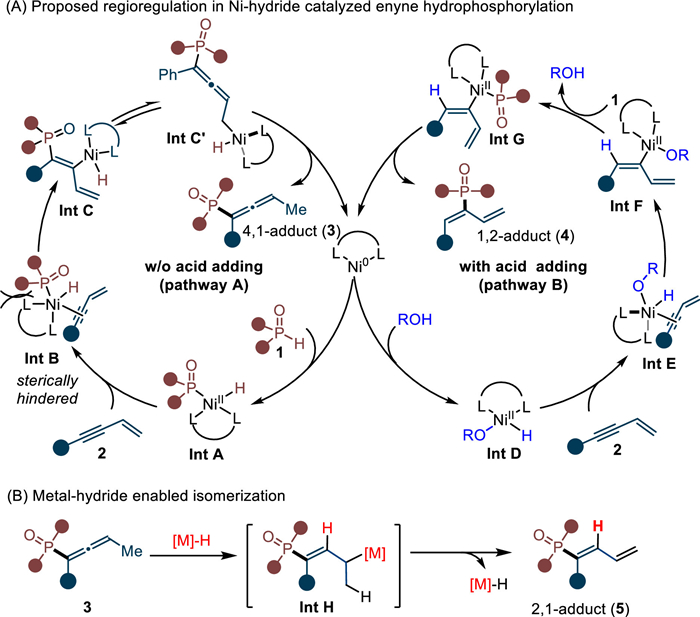
Initially, we designed this regiodivergent and atom-economic hydrophosphorylation on the basis of Ni-hydride catalysis depicted in Scheme 2A [45,61,63]. In the absence of acid, the direct oxidative addition between Ni(0) and phosphite (1) may deliver Ni-hydride species Int A, which then coordinates with enyne (2) and gives complex Int B (pathway A). Due to the bulkiness of tertiary phosphorus motif, the Int B is crowded and tends to occur P–Ni bond insertion with alkyne bond to release steric encumbrance and generate Int C or its tautomerism Int C'. Subsequently, the reductive elimination from less hindered Int C' may lead to 4,1-addition product 3 and regenerate Ni(0) species. With the aid of a Brønsted acid, the oxidative addition probably occurs first between Ni(0) and acid to yield Int D (pathway B). Next, coordination of Int D with enyne (2) furnishes complex Int E. Compared with Int B, the Int E is less crowded and Ni–H insertion with alkyne bond may take place preferentially in this case to produce Int F. Then, Int G can be obtained through the ligand exchange between phosphite (1) and Int F, followed by a final reductive elimination to deliver the 1,2-adduct 4. Notably, the 2,1-hydrophosphorylation may also be available by metal-hydride mediated alkene isomerization via Int H (Scheme 2B).

|
Download:
|
| Scheme 2. Proposed regiodivergent enyne hydrophosphorylation under Ni-hydride catalysis. | |
{kind=link}
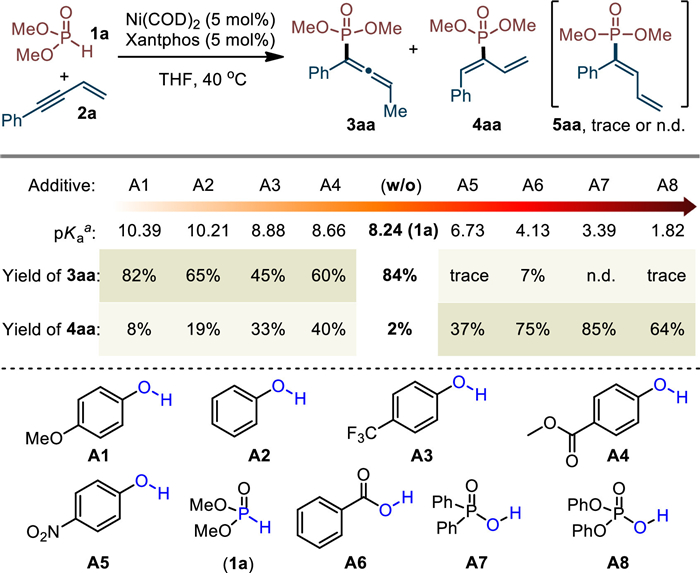
With this proposal in mind, we explored the regiodivergent hydrophosphorylation with the coupling between dimethyl phosphite (1a) and enyne (2a) and by using Ni(COD)2 as catalyst precursor in THF at 40 ℃. After the careful evaluation of various ligands (for details, see Table S1 in Supporting information), we found that Xantphos (L7) displayed best performance in terms of reactivity and the reaction could give allene 3aa in excellent selectivity. To manipulate the regioselectivity, phenols and acids with different pKa value were then examined (Scheme 3) [93]. Indeed, the electron-rich phenols with higher pKa value than 1a all gave 3aa as major product (A1 and A2). In contrast, the yield of 4aa increased while the electron-deficient phenols employed (A3 and A4) and became majority when the loaded phenol bearing pK value lower than that of dimethyl phosphite (A5 vs. 1a). These results inspired us to further screen some more acidic additives. It was found that the reaction with diphenylphosphinic acid (A7) could successfully furnish 4aa in 85% yield with excellent regioselectivity. Notably, in all case the 2,1-addition product 5aa could not be detected or only obtained in trace amount. Besides, NiCl2/Zn combined system is not suitable for current reactions (for details, see Table S2 in Supporting information).

|
Download:
|
| Scheme 3. Evaluation of reaction conditions. Conditions: 1a (0.20 mmol), 2a (0.30 mmol), Ni(COD)2 (5 mol%), Xantphos (5 mol%), additive (10 mol%), THF (0.5 mL), 40 ℃, 20 h, N2. Yield was determined by HPLC with naphthalene as the internal standard. a Predicted value with H2O as the solvent, see ref. [93]. | |
{kind=link}
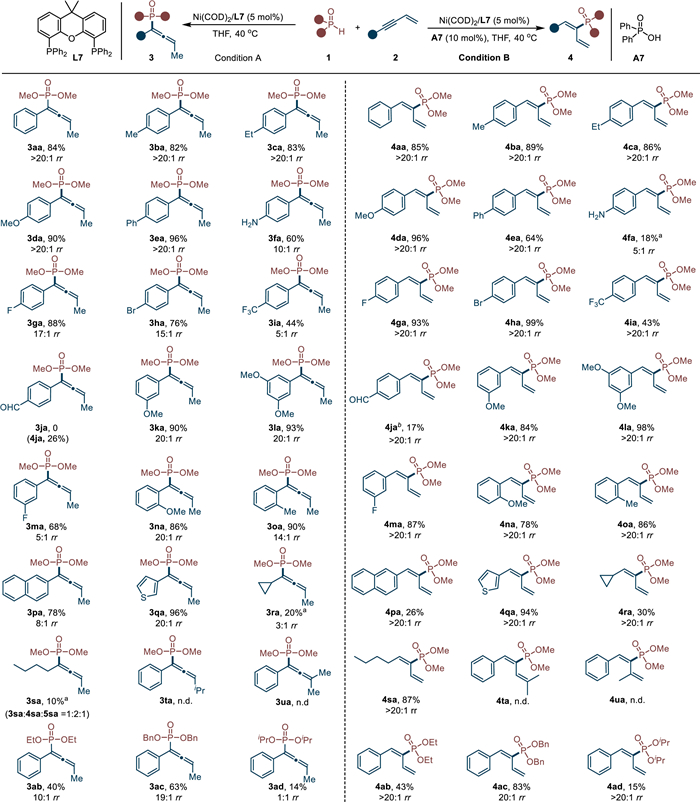
Under the standard conditions, we then sought out to examine the generality of enyne substrates for 4,1-addition (Scheme 4, condition A). A variety of enynes bearing electron-donating groups could couple with dimethyl phosphite 1a smoothly and led to allenes in good yields and regioselectivities (3aa–3fa). Halides such as -F (3ga) and -Br (3ha) were also applicable substituents for the current conversion. However, when substrates having the stronger electron-withdrawing groups, such as -CF3 (3ia) and -CHO (3ja), the reaction proceeded sluggishly. Substituents at the meta- or ortho-position of phenyl rings were compatible as well (3ka–3oa). On replacing the phenyl group with 2-naphthalenyl or 3-thienyl group, the substrates were transformed into product 3pa and 3qa in 78% and 96% yield, respectively. Aliphatic enynes could couple with 2a but resulted in low selectivity (3ra and 3sa). As illustrated by 3ta and 3ua, the reactions failed to proceed when enyne substrates bearing substituent on alkene group. When reactions were carried out with bulkier alkyl phosphites under condition A, the reactivities and regioselectivities were both decreased (3ab - 3ad). This result is probably due to steric repulsion between alkoxy group and phenyl group (Scheme 2, Int C), which makes P–Ni bond insertion step unfavorable.

|
Download:
|
| Scheme 4. Substrate scope towards 1,2-adducts and 4,1-adducts. Condition A: 1 (0.20 mmol), 2 (0.30 mmol), Ni(COD)2 (5 mol%), L7 (5 mol%), THF (0.5 mL), 40 ℃, 20 h; Condition B: 1 (0.20 mmol), 2 (0.30 mmol), Ni(COD)2 (5 mol%), L7 (5 mol%), A7 (10 mol%), THF (0.5 mL), 40 ℃, 20 h. Isolated yield of the major product was given in all cases. Regioselectivity was determined by 31P NMR analysis of the crude mixture. a Isolated product together with regioisomer, the yield of corresponding product has been adjusted accordingly. b CsOAc (10 mol%) was used instead of (Ph)2PO2H. | |
{kind=link}
With the Ni/acid catalysis, aryl enynes with various substituents at the phenyl rings successfully resulted in a range of 1,3-dienes 4aa–4pa. In most case, substrates with either electron-donating or electron-withdrawing groups all delivered products in high yields and excellent regioselectivities. However, when the enynes bearing reactive groups, such as -NH2 (4fa) and -CHO (4ja), the reactions proceeded with low yields. Delightfully, the bromo group (4ha), which is a fragile substituent in many nickel catalysis, could remain intact under current conditions and produced corresponding product in 99% yield. This reaction was also feasible to the 3-thienyl enyne and led to 4qa in 94% yield. In addition, aliphatic enyne could react with dimethyl phosphite as well, furnishing hydrophosphorylated product 4ra and 4sa in excellent selectivity. The enynes having substituent on alkene unit were also not tolerated under this condition (4ta and 4ua). In addition, the bulkiness of alkyl groups showed no obvious difference in regioselectivity (4ab - 4ad, ≥ 20:1 rr in all case). However, it was found that the bulkier alkyl group had the lower reactivity (4aa vs. 4ad). Based on the above-mentioned pathway B, the regioselectivity under acid condition was determined by the Ni–H insertion process. This step is not concerned with alkyl phosphites and therefore the regioselectivity keeps in high level. But the bulky substituents can affect the ligand exchange step, which may result in the low yield when (iPrO)P(O)H employed (4ad).
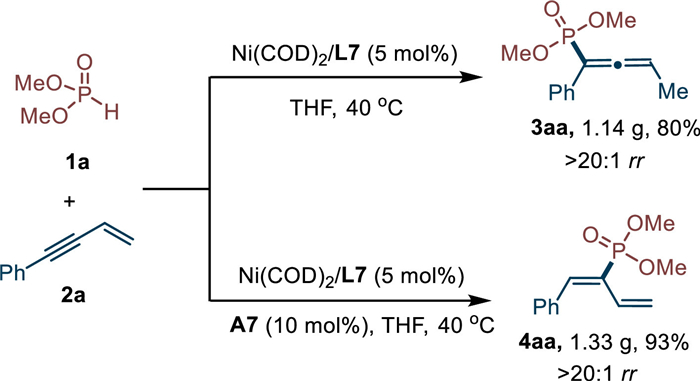
Under the optimal reaction conditions, this regiodivergent hydrophosphorylation could be easily conducted in a gram-scale (Scheme 5). Under nickel catalysis, the reaction between dimethyl phosphite (1a) and enyne (2a) could successfully deliver product 3aa in 1.14 g with 80% yield and > 20:1 rr. In the presence of acid, diene product 4aa could also be obtained in 93% yield (1.33 g) and > 20:1 rr.

|
Download:
|
| Scheme 5. Gram-scale reactions. | |
{kind=link}
To shed light on the mechanism, we also performed deuterium-labeling experiments (Scheme S1 in Supporting information). When subjecting deuterated dimethyl phosphite (1a-d) to couple with enyne (2a) under the standard condition A or condition B, respectively, it was found no obvious deuterium scrambling was detected in the products 3aa-d and 4aa-d as well as recovered 2a. These results indicate a less possibility of a reversible process between Ni−hydride insertion and β-hydride elimination. Then, kinetic studies were also performed to gain further insights on reaction profiles (Fig. S4A in Supporting information). With the increase of the reaction time, the yield of 4aa raised rapidly during the initial time under condition B, while slow linear growing was observed for 3aa under condition A. These results indicate that the reaction for 1,2-addition is rather faster than that for 4,1-addition. No transitional product being detected excludes the possibility of an interchange conversion between 3aa and 4aa.
When replacing the nickel catalyst with palladium, only 1,2-addition product was found whether acid was added or not, suggesting the unique regioregulated ability of nickel catalysis (Fig. S4B, Eqs. 1 and 2 in Supporting information). Besides, it was also found that the reaction with acid apparently exhibited higher efficiency. This is in according with kinetic experiments and previous Han's work that the adding of acid can accelerate the hydrophosphorylation process [42]. This conclusion also meets with the results when subjecting 1-phenylpropyne 6 to react with 1a under standard nickel catalysis, in which higher yields of hydrophosphorylation products (7a and 7b) were obtained under the acid condition (Fig. S4B, Eqs. 3 vs. 4). Instead, no any hydrophosphorylation product could be detected in the coupling reaction between 1-phenyl-1,3-butadiene 8 and 1a (Fig. S4B, Eq. 5), suggests that current catalysis may not favor the alkene insertion process. Moreover, diene intermediate 9 was synthesized to react with 1a under standard condition and the 6% yield of 4aa was furnished, supports the rationality of proposed pathway B (Fig. S4B, Eq. 6).
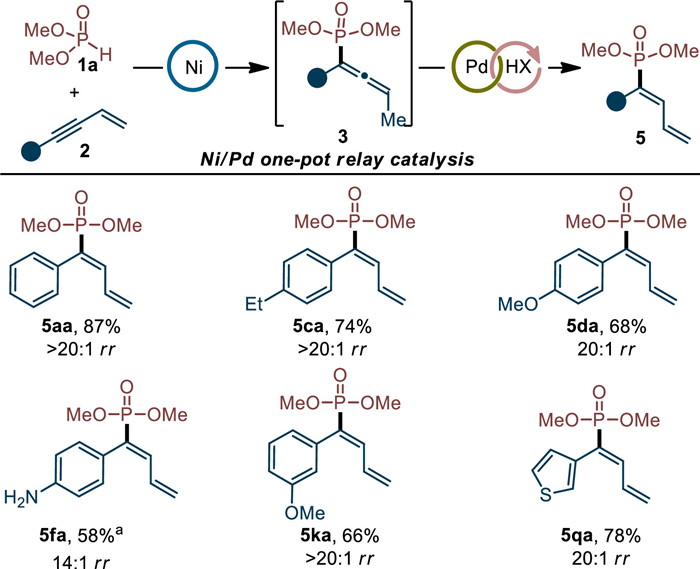
As we mentioned in Scheme 3, the selective 2,1-hydrophosphorylation of enynes was extremely difficult to be accessed by direct addition reaction. However, we conceived an indirect way—the one-pot late-stage isomerization of allene products [94,95]. With this imagination in mind, the evaluation of compatibility of Ni catalysis with isomerization catalysis was subsequently carried out (for details, see Table S5 in Supporting information). Pleasingly, it was found that under Pd(OAc)2/PPh3/PhCOOH combined catalysis, various 2,1-hydrophosphorylated products could be successfully obtained in moderate to good yields and decent regioselectivities (Scheme 6). The current relay catalysis was also featured by its easy handle and allene intermediate products were not needed to be separated in this one-pot way.

|
Download:
|
| Scheme 6. 2,1-Hydrophosphorylation by Ni/Pd one-pot relay catalysis. Step 1: 1a (0.30 mmol), 2 (0.20 mmol), Ni(COD)2 (5 mol%), L7 (5 mol%), dioxane, 40 ℃, 20 h. Step 2: Pd(OAc)2 (10 mol%), PPh3 (20 mol%), PhCOOH (40 mol%), 1,4-dioxane, 120 ℃, 6 h. Isolated yield of the major product was given in all cases. Regioselectivity was determined by 31P NMR analysis of the crude mixture. a Isolated product together with regioisomer 4fa, total yield was given. | |
{kind=link}
In conclusion, we have developed a practical strategy for the regiodivergent hydrophosphorylation of enynes. Under Ni/Xantphos catalysis, 4,1-addition could be selectively obtained whereas the adding of acid switched reactions towards 2-addition. Mechanistic studies showed that the regioselectivity was probably governed by the alkyne insertion step between (RO)2P(O)–Ni–H and (R)2P(O)O–Ni–H species. With the help of Ni/Pd relay catalysis, the 1-hydrophosphorylation could also be realized in one-pot reaction. Further investigations on more mechanistic details and the extension of this regioselectivity controlled strategy are now in progress in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support from Dalian Outstanding Young Scientific Talent (No. 2020RJ05), and the National Natural Science Foundation of China (Nos. 22071239, 21971234) is acknowledged.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.107914.
| [1] |
A. Mucha, P. Kafarski, L. Berlicki, J. Med. Chem. 54 (2011) 5955-5980. DOI:10.1021/jm200587f |
| [2] |
J.W. McGrath, J.P. Chin, J.P. Quinn, Nat. Rev. Microbiol. 11 (2013) 412-419. DOI:10.1038/nrmicro3011 |
| [3] |
S. Demkowicz, J. Rachon, M. Dasko, W. Kozak, RSC Adv. 6 (2016) 7101-7112. DOI:10.1039/C5RA25446A |
| [4] |
G.P. Horsman, D.L. Zechel, Chem. Rev. 117 (2017) 5704-5783. DOI:10.1021/acs.chemrev.6b00536 |
| [5] |
J.B. Rodriguez, C. Gallo-Rodriguez, ChemMedChem 14 (2019) 190-216. DOI:10.1002/cmdc.201800693 |
| [6] |
W.J. Tang, X.M. Zhang, Chem. Rev. 103 (2003) 3029-3069. DOI:10.1021/cr020049i |
| [7] |
H. Ni, W.L. Chan, Y. Lu, Chem. Rev. 118 (2018) 9344-9411. DOI:10.1021/acs.chemrev.8b00261 |
| [8] |
M. Mikolajczyk, P. Balczewski, Top. Curr. Chem. 223 (2003) 161-214. |
| [9] |
P. Wang, Q. Zhu, Y. Wang, et al., Chin. Chem. Lett. 32 (2021) 1432-1436. DOI:10.1016/j.cclet.2020.11.005 |
| [10] |
F.M.J. Tappe, V.T. Trepohl, M. Oestreich, Synthesis (2010) 3037-3062. |
| [11] |
I. Wauters, W. Debrouwer, C.V. Stevens, Beilstein J. Org. Chem. 10 (2014) 1064-1096. DOI:10.3762/bjoc.10.106 |
| [12] |
Z.Y. Wang, Q. Guo, S. Xu, K.K. Wang, Synthesis 53 (2021) 3683-3698. DOI:10.1055/a-1511-0382 |
| [13] |
J. Yuan, W.P. To, Z.Y. Zhang, et al., Org. Lett. 20 (2018) 7816-7820. DOI:10.1021/acs.orglett.8b03265 |
| [14] |
J.Q. Buquoi, J.M. Lear, X. Gu, D.A. Nagib, ACS Catal. 9 (2019) 5330-5335. DOI:10.1021/acscatal.9b01580 |
| [15] |
Q. Dai, W. Li, Z. Li, J. Zhang, J. Am. Chem. Soc. 141 (2019) 20556-20564. DOI:10.1021/jacs.9b11938 |
| [16] |
C. Liu, C.L. Ji, T. Zhou, X. Hong, M. Szostak, Org. Lett. 21 (2019) 9256-9261. DOI:10.1021/acs.orglett.9b03678 |
| [17] |
Y. Liu, P. Xie, J. Li, W.J. Bai, J. Jiang, Org. Lett. 21 (2019) 4944-4949. DOI:10.1021/acs.orglett.9b01288 |
| [18] |
H.Q. Cao, H.N. Liu, Z.Y. Liu, et al., Org. Lett. 22 (2020) 6414-6419. DOI:10.1021/acs.orglett.0c02229 |
| [19] |
J.S. Zhang, T. Chen, L.B. Han, Eur. J. Org. Chem. 2020 (2020) 1148-1153. DOI:10.1002/ejoc.201901865 |
| [20] |
Y. Song, L. Wang, Z. Duan, F. Mathey, Chin. Chem. Lett. 31 (2020) 329-332. DOI:10.1016/j.cclet.2019.05.053 |
| [21] |
W. Yang, B. Li, M. Zhang, et al., Chin. Chem. Lett. 31 (2020) 1313-1316. DOI:10.1016/j.cclet.2019.10.022 |
| [22] |
H. Long, C. Huang, Y.T. Zheng, et al., Nat. Commun. 12 (2021) 6629. DOI:10.1038/s41467-021-26960-y |
| [23] |
Z.K. Tao, C.K. Li, J.A. Li, et al., Org. Lett. 23 (2021) 4342-4347. DOI:10.1021/acs.orglett.1c01286 |
| [24] |
S. Wang, Q. Xue, Z. Guan, Y. Ye, A. Lei, ACS Catal. 11 (2021) 4295-4300. DOI:10.1021/acscatal.1c00549 |
| [25] |
B. Li, M. Liu, S.U. Rehman, C. Li, J. Am. Chem. Soc. 144 (2022) 2893-2898. DOI:10.1021/jacs.2c00239 |
| [26] |
L. Coudray, J.L. Montchamp, Eur. J. Org. Chem. 2008 (2008) 3601-3613. DOI:10.1002/ejoc.200800331 |
| [27] |
D.P. Zhao, R. Wang, Chem. Soc. Rev. 41 (2012) 2095-2108. DOI:10.1039/C1CS15247E |
| [28] |
L. Rosenberg, ACS Catal. 3 (2013) 2845-2855. DOI:10.1021/cs400685c |
| [29] |
V. Koshti, S. Gaikwad, S.H. Chikkali, Coord. Chem. Rev. 265 (2014) 52-73. DOI:10.1016/j.ccr.2014.01.006 |
| [30] |
C.A. Bange, R. Waterman, Chem. Eur. J. 22 (2016) 12598-12605. DOI:10.1002/chem.201602749 |
| [31] |
T. Xiong, H. Yuan, F. Yang, J. Jiang, Green Synth. Catal. 3 (2022) 46-52. DOI:10.1016/j.gresc.2021.10.005 |
| [32] |
W.Q. Liu, T. Lei, S. Zhou, et al., J. Am. Chem. Soc. 141 (2019) 13941-13947. DOI:10.1021/jacs.9b06920 |
| [33] |
Z. Lu, H. Zhang, Z. Yang, et al., ACS Catal. 9 (2019) 1457-1463. DOI:10.1021/acscatal.8b04787 |
| [34] |
Y.A. Rina, J.A.R. Schmidt, Organometallics 38 (2019) 4261-4270. DOI:10.1021/acs.organomet.9b00549 |
| [35] |
W.J. Yue, J.Z. Xiao, S. Zhang, L. Yin, Angew. Chem. Int. Ed. 59 (2020) 7057-7062. DOI:10.1002/anie.201916076 |
| [36] |
C. Wang, K. Huang, J. Ye, W.L. Duan, J. Am. Chem. Soc. 143 (2021) 5685-5690. DOI:10.1021/jacs.1c02772 |
| [37] |
L. Ge, S.R. Harutyunyan, Chem. Sci. 13 (2022) 1307-1312. DOI:10.1039/D1SC06694C |
| [38] |
L. Huang, E.Q. Lim, M.J. Koh, Chem. Catal. 2 (2022) 508-518. DOI:10.1016/j.checat.2021.12.014 |
| [39] |
L.B. Han, N. Choi, M. Tanaka, Organometallics 15 (1996) 3259-3261. DOI:10.1021/om960328u |
| [40] |
L.B. Han, M. Tanaka, J. Am. Chem. Soc. 118 (1996) 1571-1572. DOI:10.1021/ja953690t |
| [41] |
L.B. Han, R.M. Hua, M. Tanaka, Angew. Chem. Int. Ed. 37 (1998) 94-96. DOI:10.1002/(SICI)1521-3773(19980202)37:1/2<94::AID-ANIE94>3.0.CO;2-T |
| [42] |
L.B. Han, F. Mirzaei, C.Q. Zhao, M. Tanaka, J. Am. Chem. Soc. 122 (2000) 5407-5408. DOI:10.1021/ja000444v |
| [43] |
C.Q. Zhao, L.B. Han, M. Goto, M. Tanaka, Angew. Chem. Int. Ed. 40 (2001) 1929-1932. DOI:10.1002/1521-3773(20010518)40:10<1929::AID-ANIE1929>3.0.CO;2-M |
| [44] |
L.B. Han, C.Q. Zhao, S.Y. Onozawa, M. Goto, M. Tanaka, J. Am. Chem. Soc. 124 (2002) 3842-3843. DOI:10.1021/ja025816+ |
| [45] |
L.B. Han, C. Zhang, H. Yazawa, S. Shimada, J. Am. Chem. Soc. 126 (2004) 5080-5081. DOI:10.1021/ja0494297 |
| [46] |
P. Ribiere, K. Bravo-Altamirano, M.I. Antczak, J.D. Hawkins, J.L. Montchamp, J. Org. Chem. 70 (2005) 4064-4072. DOI:10.1021/jo050096l |
| [47] |
T. Chen, C.Q. Zhao, L.B. Han, J. Am. Chem. Soc. 140 (2018) 3139-3155. DOI:10.1021/jacs.8b00550 |
| [48] |
H. Wang, Y. Li, Z. Tang, et al., ACS Catal. 8 (2018) 10599-10605. DOI:10.1021/acscatal.8b02617 |
| [49] |
F. Chen, Y. Xia, R. Lin, et al., Org. Lett. 21 (2019) 579-583. DOI:10.1021/acs.orglett.8b03985 |
| [50] |
Q. Dai, L. Liu, Y. Qian, W. Li, J. Zhang, Angew. Chem. Int. Ed. 59 (2020) 20645-20650. DOI:10.1002/anie.202009358 |
| [51] |
D.S. Glueck, J. Org. Chem. 85 (2020) 14276-14285. DOI:10.1021/acs.joc.0c00667 |
| [52] |
Z. Yang, X. Gu, L.B. Han, J. Wang, Chem. Sci. 11 (2020) 7451-7455. DOI:10.1039/D0SC01049A |
| [53] |
X.T. Liu, X.Y. Han, Y. Wu, et al., J. Am. Chem. Soc. 143 (2021) 11309-11316. DOI:10.1021/jacs.1c05649 |
| [54] |
S. Hu, W. Sun, J. Chen, et al., Chem. Commun. 57 (2021) 339-342. DOI:10.1039/D0CC07022J |
| [55] |
Z. Yang, J. Wang, Angew. Chem. Int. Ed. 60 (2021) 27288-27292. DOI:10.1002/anie.202112285 |
| [56] |
S. Kwak, J. Choi, J. Han, S.Y. Lee, ACS Catal. 12 (2022) 212-218. DOI:10.1021/acscatal.1c04242 |
| [57] |
T. Hirao, T. Masunaga, N. Yamada, Y. Ohshiro, T. Agawa, Bull. Chem. Soc. Jpn. 55 (1982) 909-913. DOI:10.1246/bcsj.55.909 |
| [58] |
F. Mirzaei, L.B. Han, M. Tanaka, Tetrahedron Lett. 42 (2001) 297-299. DOI:10.1016/S0040-4039(00)01928-6 |
| [59] |
X.Y. Yang, W.S. Tay, Y. Li, S.A. Pullarkat, P.H. Leung, Organometallics 34 (2015) 5196-5201. DOI:10.1021/acs.organomet.5b00787 |
| [60] |
S.Z. Nie, R.T. Davison, V.M. Dong, J. Am. Chem. Soc. 140 (2018) 16450-16454. DOI:10.1021/jacs.8b11150 |
| [61] |
J. Long, Y. Li, W. Zhao, G. Yin, Chem. Sci. 13 (2022) 1390-1397. DOI:10.1039/D1SC05651D |
| [62] |
H. Hou, B. Zhou, J. Wang, et al., Org. Lett. 23 (2021) 2981-2987. DOI:10.1021/acs.orglett.1c00626 |
| [63] |
Y.Q. Zhang, X.Y. Han, Y. Wu, et al., Chem. Sci. 13 (2022) 4095-4102. DOI:10.1039/D2SC00091A |
| [64] |
M. Holmes, L.A. Schwartz, M.J. Krische, Chem. Rev. 118 (2018) 6026-6052. DOI:10.1021/acs.chemrev.8b00213 |
| [65] |
J. Chen, J. Guo, Z. Lu, Chin. J. Chem. 36 (2018) 1075-1109. DOI:10.1002/cjoc.201800314 |
| [66] |
Q. Dherbassy, S. Manna, F.J.T. Talbot, et al., Chem. Sci. 11 (2020) 11380-11393. DOI:10.1039/D0SC04012F |
| [67] |
L. Fu, S. Gressies, P. Chen, G. Liu, Chin. J. Chem. 38 (2020) 91-100. DOI:10.1002/cjoc.201900277 |
| [68] |
H.Y. Jang, R.R. Huddleston, M.J. Krische, J. Am. Chem. Soc. 126 (2004) 4664-4668. DOI:10.1021/ja0316566 |
| [69] |
V. Komanduri, M.J. Krische, J. Am. Chem. Soc. 128 (2006) 16448-16449. DOI:10.1021/ja0673027 |
| [70] |
R.L. Patman, V.M. Williams, J.F. Bower, M.J. Krische, Angew. Chem. Int. Ed. 47 (2008) 5220-5223. DOI:10.1002/anie.200801359 |
| [71] |
T. Nishimura, H. Makino, M. Nagaosa, T. Hayashi, J. Am. Chem. Soc. 132 (2010) 12865-12867. DOI:10.1021/ja1066509 |
| [72] |
K.D. Nguyen, D. Herkommer, M.J. Krische, J. Am. Chem. Soc. 138 (2016) 5238-5241. DOI:10.1021/jacs.6b02279 |
| [73] |
Y. Yang, I.B. Perry, G. Lu, P. Liu, S.L. Buchwald, Science 353 (2016) 144-150. DOI:10.1126/science.aaf7720 |
| [74] |
S. Yu, H.L. Sang, S.Q. Zhang, X. Hong, S. Ge, Commun. Chem. 1 (2018) 64. DOI:10.1038/s42004-018-0065-4 |
| [75] |
C. Law, E. Kativhu, J. Wang, J.P. Morken, Angew. Chem. Int. Ed. 59 (2020) 10311-10315. DOI:10.1002/anie.202001580 |
| [76] |
S.Q. Yang, Y.F. Wang, W.C. Zhao, G.Q. Lin, Z.T. He, J. Am. Chem. Soc. 143 (2021) 7285-7291. DOI:10.1021/jacs.1c03157 |
| [77] |
U. Radhakrishnan, M. Al-Masum, Y. Yamamoto, Tetrahedron Lett. 39 (1998) 1037-1040. DOI:10.1016/S0040-4039(97)10697-9 |
| [78] |
C. Brinkmann, A.G.M. Barrett, M.S. Hill, P.A. Procopiou, J. Am. Chem. Soc. 134 (2012) 2193-2207. DOI:10.1021/ja209135t |
| [79] |
N.J. Adamson, H. Jeddi, S.J. Malcolmson, J. Am. Chem. Soc. 141 (2019) 8574-8583. DOI:10.1021/jacs.9b02637 |
| [80] |
J.W. Han, N. Tokunaga, T. Hayashi, J. Am. Chem. Soc. 123 (2001) 12915-12916. DOI:10.1021/ja017138h |
| [81] |
W. Chen, C. Jiang, J. Zhang, et al., J. Am. Chem. Soc. 143 (2021) 12913-12918. DOI:10.1021/jacs.1c04689 |
| [82] |
D.W. Gao, Y. Xiao, M. Liu, et al., ACS Catal. 8 (2018) 3650-3654. DOI:10.1021/acscatal.8b00626 |
| [83] |
Y. Huang, J. del Pozo, S. Torker, A.H. Hoveyda, J. Am. Chem. Soc. 140 (2018) 2643-2655. DOI:10.1021/jacs.7b13296 |
| [84] |
H.L. Sang, S. Yu, S. Ge, Org. Chem. Front. 5 (2018) 1284-1287. DOI:10.1039/C8QO00167G |
| [85] |
F.F. Meng, Q.Y. Xue, B. Jiang, et al., Org. Lett. 21 (2019) 2932-2936. DOI:10.1021/acs.orglett.9b00995 |
| [86] |
C. Yang, Z.L. Liu, D.T. Dai, et al., Org. Lett. 22 (2020) 1360-1367. DOI:10.1021/acs.orglett.9b04647 |
| [87] |
Y.C. Hu, D.W. Ji, C.Y. Zhao, H. Zheng, Q.A. Chen, Angew. Chem. Int. Ed. 58 (2019) 5438-5442. DOI:10.1002/anie.201901025 |
| [88] |
D.W. Ji, Y.C. Hu, H. Zheng, et al., Chem. Sci. 10 (2019) 6311-6315. DOI:10.1039/C9SC01527B |
| [89] |
C.S. Kuai, D.W. Ji, C.Y. Zhao, et al., Angew. Chem. Int. Ed. 59 (2020) 19115-19120. DOI:10.1002/anie.202007930 |
| [90] |
W.S. Jiang, D.W. Ji, W.S. Zhang, et al., Angew. Chem. Int. Ed. 60 (2021) 8321-8328. DOI:10.1002/anie.202100137 |
| [91] |
X. Huang, B.Z. Chen, P. Li, et al., Nature Chem. 14 (2022) 1185-1192. DOI:10.1038/s41557-022-01017-9 |
| [92] |
G. Zhang, C.Y. Zhao, X.T. Min, et al., Nature Catal. 5 (2022) 708-715. DOI:10.1038/s41929-022-00825-z |
| [93] |
Q. Yang, Y. Li, J.D. Yang, et al., Angew. Chem. Int. Ed. 59 (2020) 19282-19291. DOI:10.1002/anie.202008528 |
| [94] |
M. Al-Masum, Y. Yamamoto, J. Am. Chem. Soc. 120 (1998) 3809-3810. DOI:10.1021/ja974223+ |
| [95] |
Y. Al-Jawaheri, M.C. Kimber, Org. Lett. 18 (2016) 3502-3505. DOI:10.1021/acs.orglett.6b01841 |