 2023, Vol. 34
2023, Vol. 34 


b Neutron Scattering Division, Oka Ridge National Laboratory, Oak Ridge 37831, United States;
c Guangxi Key Laboratory of Electrochemical and Magnetochemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, China
Silicates are known for their natural abundance and are thus widely used in domestic ceramics, glassware, refractories, and building and construction materials [1,2]. Apart from these applications as structural materials, silicates also display interesting functional properties including ferroelectricity [3], nonlinear optical effects [4], luminescence host [5], and ionic conduction [6]. With a variety of chemical composition and structural variations, silicates have the tetrahedral SiO44− anion with relative rigidity and strong covalent interactions as the basic structural feature under ambient pressure (AP). The SiO44− anions could be isolated [7], or connected into small polymers [8,9], one-dimensional (1D) chains [10], two-dimensional (2D) layers [11], and three-dimensional (3D) framework [12] via apex-oxygen sharing, hosting a great variety of metal cations in the resulted voids to compensate the charge.
The ionic metal oxide usually adopts a close packing arrangement of oxygen (and large-size cations) with the metal cations filling in the resulted voids with oxygen coordination number (CN) ≥ 6, typically BO6 octahedron and AO12 truncated octahedron in ABO3 perovskites [13]. In contrast to the ionic metal oxide case, the oxygen ions in the silicates are loosely stacked to satisfy the tetrahedral coordination requirement of Si4+, leading to low space utilization and small CN for the metal cations. For example, typical sodium silicates Na2SiO3 [14] and α-Na2Si2O5 [15] have 5-coordinated Na+ cations, while the other ternary ionic metal oxides normally have 6-12 coordinated environments for Na+, such as those in NaMnO2 [16] and NaNbO3 [17].
Applying high pressures would alter the loosely anionic stacking of the silicates and lead to structural modifications. For example, the stable phase of MgSiO3 enstatite [18] at ambient conditions contains alternative layers of MgO6 octahedron and SiO4 tetrahedra (Fig. S1a in Supporting information), which would transform into MgSiO3 ilmenite (Fig. S1b in Supporting information) with SiO6 octahedral layers at 20-24 GPa (1373-2273 K) [19,20]. Further increasing pressure above 23 GPa (T > 1473 K) leads to the stabilization of MgSiO3 perovskite (Fig. S1c in Supporting information) [21,22], which would convert into the post-perovskite CaIrO3 structure with edge-sharing SiO6 octahedral chains at pressures > 125 GPa (T > 2500 K) (Fig. S1d in Supporting information) [23]. Due to the different initial size contrast between the ions and atomic compressibility of elements, the structural variation with pressure are different when the cations are changed, as shown by that CaSiO3 parawollastonite [10] with 1D SiO32− chains (Fig. S1e in Supporting information) can also transform into perovskite at 14-16 GPa [24] and SrSiO3, which are composed of alternating layers of Si3O96− rings and layers of 8-coordinated Sr atoms (Fig. S1f in Supporting information) [9], convert into hexagonal perovskite at 20 GPa then cubic perovskite at 32 GPa [25,26]. Generally, the non-perovskite silicates MSiO3 with small B-site cations would adopt perovskite structure at certain pressure as those observed in other ABO3 systems with small A-site cations [27].
Recently lanthanum silicates have been explored extensively under AP for their interesting properties on luminescence and ionic conduction, e.g., La2SiO5 [28], La9.33+x(SiO4)6O2+1.5x (x = 0-0.67) [29,30] and La2Si2O7 [8,31,32]. La2SiO5 features an isolated SiO4 tetrahedron and oxosilicate nature with some oxygen anions only bonding to La cations (Fig. S1g in Supporting information). While La2Si2O7 has four polymorphs: the A-type (tetragonal, P41), G-type (monoclinic, P21/c), and H-type (triclinic, P1) structures containing diorthosilicate Si2O76− anions built up of two corner-linked SiO4 tetrahedra (Fig. S1h in Supporting information) [8,31], the I-type containing a hoof-shaped catena-tetrasilicate Si4O1310− ions and two orthosilicates SiO44− ions per formula unit (Fig. S1i in Supporting information) [32]. Among these four polymorphs of La2Si2O7, the tetragonal A-type phase is thermodynamically stable at room temperature, while the other phases are metastable [8]. The La9.33+x(SiO4)6O2+1.5x (x = 0-0.67) compositions, which lie between La2SiO5 and La2Si2O7, adopt apatite structure, which is built up of isolated SiO4 tetrahedron with La3+ cations located in the 7- and 9-coordinated sites and the remaining O2− anions occupying channel along the c-axis (Fig. S1j in Supporting information). The La9.33+x(SiO4)6O2+1.5x apatite features extra oxygen atoms or La vacancies, which prompt oxide conduction through an interstitial mechanism [29,33]. Replacing La partially with Sr in La9.33+x(SiO4)6O2+1.5x leads to a stoichiometric apatite of La8Sr2(SiO4)6O2 with the oxide ion mobility reduced significantly [34]. Compared with the alkaline earth silicates, the lanthanum-based silicates received less attention on the pressure-induced structural evolution. A high-pressure (HP) lanthanum silicate has been reported as La4Si3O12 with a monoclinic cell twenty years ago. However, the crystal structure of the HP silicate La4Si3O12 is problematic and contains only rudimentary information [35,36].
For the non-perovskite ABO3 compositions under AP, it is relatively easy to predicate the HP transformation to perovskite structure although there could be intermediate polymorphs before reaching the dense perovskite structure. While for the compositions highly deviated from ABO3, the prediction of transformation to perovskite structure is not straightforward under high pressure. However, the defected perovskite structures could be expected if these compositions can form perovskite-related structures under high pressure, which may provide an effective strategy for new functional materials. So far there is little attention to HP preparation of the defected perovskite-related structures from the compositions highly deviated from ABO3 [37].
To address the possibility of perovskite-related structure formation in the compositions highly deviated from ABO3, we have been investigating the structural evolution of the less explored lanthanum silicates under high pressure. Herein we report that La6Sr3Si6O24 composition, which forms a mixed apatite and SrSiO3 phase at ambient pressure, transformed to a highly defected hexagonal perovskite-related structure with both B-cation and oxygen deficiencies under high pressure around 6 GPa. This hexagonal perovskite-related structure features B-cation and oxygen vacancy ordering, leading to isolated tetrahedral SiO4 units, and ionic-conducting behavior.
The synthesis attempt at the composition of La6Sr3Si6O24 at 1473 K and AP leads to a mixed phase of La8Sr2Si6O26 apatite [34] and SrSiO3 [9], as evidenced by the Rietveld plot shown in Fig. S2a (Supporting information). The coexistence of two phases with significant compositional contrast is directly observed with SEM images from backscattered electrons and EDS elemental mapping, as shown in Fig. S3 (Supporting information). An unknown HP phase started to appear on the XRD data of La6Sr3Si6O24 (Fig. S2b in Supporting information) as the applied pressure reached 3 GPa and above, and its phase content increased with pressure. At pressure above 6 GPa, the reflections of the AP phases disappeared, and the HP phase was isolated as a single-phase material. The EDS elemental analysis of the HP material gives an average cation ratio of La6.2(4)Sr3Si6.1(2) across the sample (Figs. S4 and S5 in Supporting information), which reasonably keeps with the nominal stoichiometric composition of La6Si3Si6O24.
The XRD pattern of HP La6Sr3Si6O24 sample can be indexed into a hexagonal cell with lattice parameters of a = 5.4326(8) Å and c = 19.6174(3) Å, showing the reflection conditions: hkil: −h + k + l = 3n and 000l: l = 3n, which suggest a rhombohedral hexagonal lattice type and possible space groups of R3, R3, R32, R3m, and R3m. Figs. 1a and b show the SAED patterns of the La6Sr3Si6O24 sample collected along the [100] and [001] directions, respectively. No superstructure weak reflections were observed and all the reflections are compatible with the hexagonal cells and the possible space groups above. Therefore, the space group R3m with the highest symmetry was chosen in the following structure determination.

|
Download:
|
| Fig. 1. Electron diffraction patterns of La6Sr3Si6O24 recorded along the (a) [100] and (b) [001] directions. | |
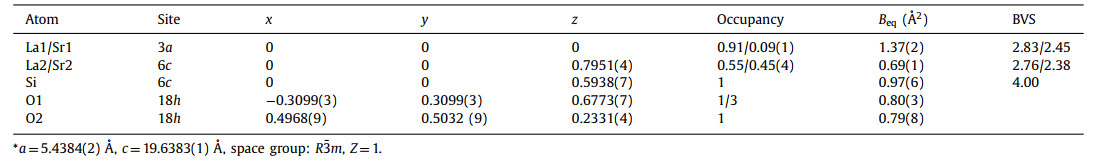
The ab initio structure solution of La6Sr3Si6O24 was performed based on the XRD data in the space group R3m by the direct method using EXPO2014 [38,39], in which the initial positions of heavier lanthanum and strontium atoms can be directly determined. Subsequently, silicon and oxygen positions were obtained by the difference Fourier analysis. Rietveld refinements of lattice parameters, atomic positions, and isotropic displacement parameters (Biso) were carried out simultaneously against the XRD and NPD data. The mixed occupancies of La and Sr were identified and refined on the 10 and 12-fold coordinated sites. The initial refinement found a relatively large atomic displacement parameter on the ideal O1 site 6c (0, 0, z). Therefore, a split general position 18h with a fractional occupancy of one third was adopted for the O1 site in the following refinements. The final refinement converged to reliability factors Rwp of ~4.62% and 6.85% for XRD and NPD data as shown in Figs. 2a and b, respectively, and the final refined atomic coordinates are given in Table 1.

|
Download:
|
| Fig. 2. Observed (red crosses), calculated (green line) and difference plots (black line) for the structure refinement of HP La6Sr3Si6O24 against (a) XRD and (b) NPD data. The Rwp (Rp) factors are ~6.17% (~7.29%) and ~4.62% (~3.49%) for NPD and XRD data, respectively. The inset in (a) enlarges the reflections in the 2θ range from 18° to 36°. | |
|
|
Table 1 Final refined structural parameters for La6Sr3Si6O24*. |
{kind=link}
{kind=link}
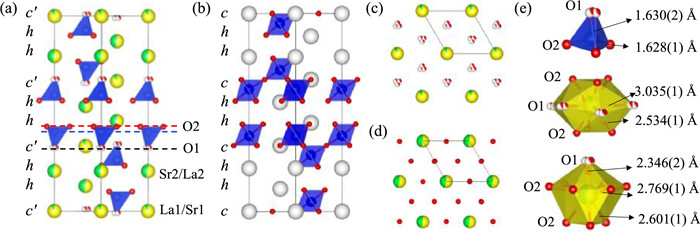
The crystal structure of HP La6Sr3Si6O24 can be described as both oxygen and B-cation deficient hexagonal perovskite (Fig. 3a), in comparison with its corresponding 9-layer B-site deficient shifted hexagonal perovskite A3B2O9 [40] with the similar stacking sequence of (chh)3 (c and h denote cubic and hexagonal stacking of close-packed AO3 layers, respectively) and oxygen vacancies filled (Fig. 3b). The stacking sequence of HP La6Sr3Si6O24 is (c′hh)3, where c′ denotes an oxygen-deficient (La/Sr)O2 pseudo-cubic-stacking layer (Fig. 3c), and h denotes a close-packed (La/Sr)O3 layer (Fig. 3d). Due to the formation of the (La/Sr)O2 oxygen-deficient layer and oxygen vacancy ordering, the 6-coordinated octahedral site formed between the neighboring c-layer and h-layer in A3B2O9 perovskite turned into the 4-coordinated tetrahedral site, which is occupied by the perovskite B-site cations (here, Si4+ cation). The octahedral sites between the two adjacent h layers are vacant in La6Sr3Si6O24, leading to a layered arrangement of B-site Si4+ cations tetrahedra and octahedral vacancies. The SiO4 tetrahedron has two sets of Si-O bonds with essentially identical lengths of ~1.63 Å (Fig. 3e top), which keeps with its average value of ~1.62 Å in the literature [1].

|
Download:
|
| Fig. 3. The structure viewed along the [110] direction for (a) La6Sr3Si6O24 oxygen-deficient perovskite and (b) its corresponding 9-layer shifted hexagonal perovskite A3B2O9 without oxygen vacancy. (c) and (d) show the oxygen-deficient c'-(La/Sr)O2 layer and the close-packed h-(La/Sr)O3 layer in La6Sr3Si6O24, as marked with dashed black and red lines in (a), respectively. (e) The coordination environments of Si, La1/Sr1, and Sr2/La2 from the top to bottom. | |
{kind=link}
Interestingly, the A-site cations La and Sr in La6Sr3Si6O24 are not randomly distributed over the 3a and 6c sites in the c′ and h layers although they have a similar ionic radius of 1.44 Å and 1.36 Å (with CN = 12), respectively [41]. This partial site occupancy leads to relatively low bond valence sum (BVS) values of La3+ (2.83 and 2.76) and high BVS values for Sr2+ (2.45 and 2.38) on both sites, similar to those in La8Sr2Si6O26 [34]. The La atoms prefer to occupy the 3a site in the oxygen-deficient AO2 layer, forming a 12-coordinated environment (Fig. 3e middle), while the Sr cations concentrate in the close-packed AO3 layer with CN = 10 (Fig. 3e bottom). This site selectivity is probably driven by the charge difference rather than the size difference since the slightly larger Sr2+ cation usually prefers a higher CN than La3+ ion. As shown in Fig. 3a, the Si4+ cationic layer (dashed blue line) is much closer to the h-AO3 layer (red dashed line) than the c′-AO2 layer (dashed black line), and has dominant electrostatic interaction with the former. To balance the positive charge of the adjacent Si4+ layer, the h-AO3 layers prefer to accommodate nearly all available Sr2+ than La3+ ions to carry more negative charges.
The thermal stability of La6Sr3Si6O24 was investigated by the thermal analysis (Fig. S6a in Supporting information) and temperature varied XRD (Fig. S6b in Supporting information). No phase transition was observed on the in-situ XRD data within the 298-1073 K temperature range, while trace amounts of impurity reflections appeared at T = 1273 K, indicative of decomposition. A complete decomposition was reached for the sample heated at 1273 K for 12 h (Fig. S6c in Supporting information). The decomposition started below 1173 K, as suggested by the secondary peaks on the XRD plot of the sample annealed at 1173 K for 6 h. No impurity phases were observed for the HP La6Sr3Si6O24 compound annealed at 1073 K for 10 h, suggesting its good dynamical stability. The lattice parameters of HP La6Sr3Si6O24 phase increase linearly as the temperature increases (Fig. S6d in Supporting information), showing anisotropic thermal expansions of αa = 11.7 × 10−6 K−1 and αc = 7.9 × 10−6 K−1, keeping with rhombohedral structural anisotropy. This leads to an overall linear volume expansion of αv = 31.6 × 10−6 K−1 at the 298-1273 K temperature range.
Generally, the application of high pressure tends to densify the materials by leading to a shorter atomic distance and thus increasing the CN of the cation and anions [42,43]. The denser atomic stacking in the HP La6Sr3Si6O24 phase can be understood through the structure comparison with the AP SrSiO3, La8Sr2Si6O26 phases, in which although the Si atoms have similar 4-fold coordinations, the La and Sr atoms have different coordination environments. The Sr2+ cations are 8-oxygen coordinated in the SrSiO3 phase, in contrast with the 7 and 9-oxygen coordinated La3+ and Sr2+ ions in the La8Sr2Si6O26 apatite. However, the HP La6Sr3Si6O24 phase has larger CN for Sr and La than both AP phases, i.e., 12 and 10. Thus, the driving force for the phase reaction of 6SrSiO3 + 3La8Sr2Si6O26 → 4La6Sr3Si6O26 can be related to the increasing of the cationic CN and the more close-packed cations and anions under pressure.
To explore the transport properties of La6Sr3Si6O24, AC impedance data was collected on the 1027 K annealed pellet, which has 95.4(1)% of the theoretical density. The samples as-quenched from the HP synthesis have many strain-induced defects and a large proportion of small grains (Fig. S4), which would recrystallize during the measurement, evidenced by the exothermal thermal peak at ~600 K (Fig. S6a) and give history-dependent impedance data. However, the La6Sr3Si6O24 sample annealed at 1073 K contains large grains and well-defined grain boundaries (Fig. S5) and gives identical conductivities on heating and cooling.
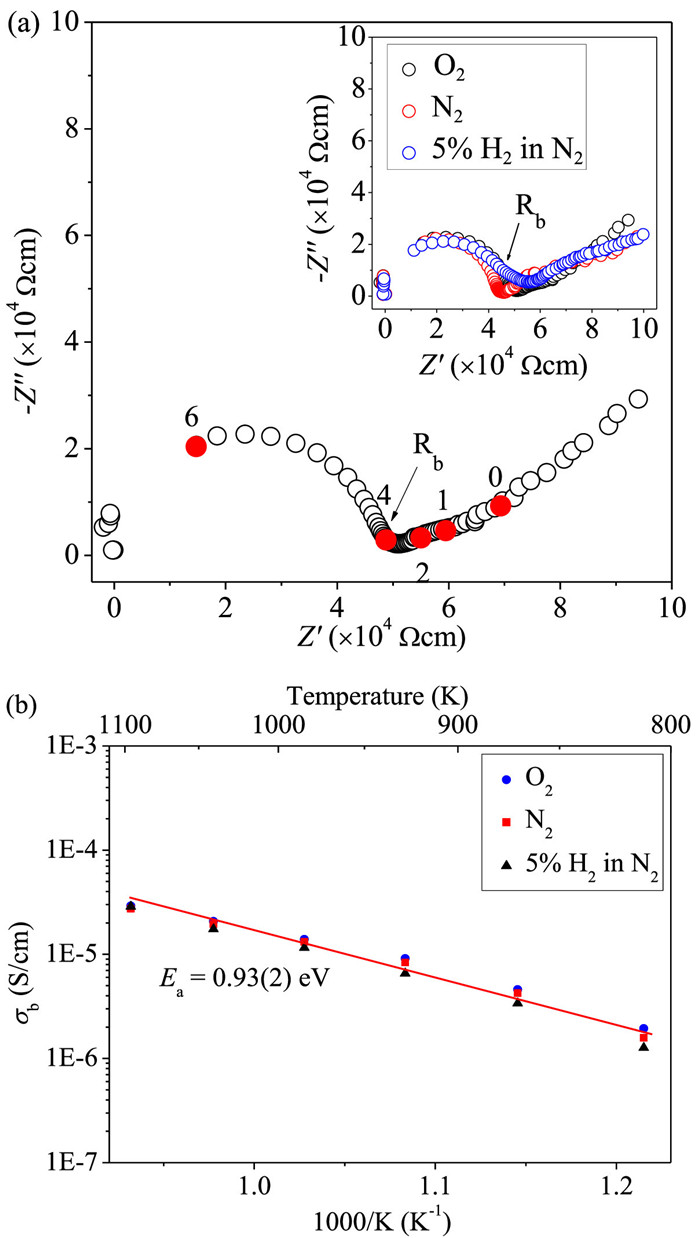
Fig. 4a shows typical complex impedance plots at 1023 K measured at different atmospheres, which are essentially identical and consist of one symmetric semicircle arc and apparent Warburg-type electrode response tails. The semicircular arc can be modeled with a parallel Resistor-Capacitance (RC) circuit: the intercept of the semicircular arc at low frequency is estimated as R; the associated C value calculated from the equation 2πfmaxRC = 1 (fmax is the frequency corresponding to the maximum imaginary impedance Zmax′′) is ~4.9 pF/cm. This value is close to that (~5.4 pF/cm) estimated from the maximum imaginary modulus Mmax′′ using the equation ε0/(2Mmax′′) (ε0 is the capacitance of free space, 8.854 × 10−14 F/cm). This confirms that the semicircular arc can be ascribed to a single bulk component response [44,45]. The electrode responses in the impedance data of La6Sr3Si6O24 pellet show large capacitances of ~10−8-10−6 F/cm in the 10-0.1 Hz low-frequency region, which is indicative of ionic conduction in La6Sr3Si6O24 [45]. Apart from the semicircular-arc bulk response and the inclined-line electrode response, no apparent semicircular arc was observed from the grain boundary response, which however could be disguised between the bulk and electrode responses as indicated by the capacitances around 10−11-10−9 F/cm in the 104-102 Hz frequency range.

|
Download:
|
| Fig. 4. (a) The 1023 K complex impedance plots for La6Sr3Si6O24 pellets measured under O2 atmosphere in comparison with those in N2 and 5% H2 in N2 atmospheres in the inset. Rb denotes the bulk resistivity and the numbers denote the logarithms of selected frequencies marked by filled symbols. (b) Temperature-dependent bulk conductivities of La6Sr3Si6O24 pellets in the O2, N2, and 5% H2 in N2 atmospheres with activation energy (Ea) labeled. | |
{kind=link}
Recently the B-cation and oxygen-deficient hexagonal perovskite transitional metal oxides (e.g., Ba3WNbO8.5 [46], Ba7Nb4MoO20 [47,48], and Ba7Y2Mn3Ti2O20 [49]) have been shown to transport the oxide ions. In these hexagonal perovskites, the oxygen-vacancy ordered c'-AO2 layers could transform to oxygen-vacancy disordered c-AO3−x layers thus allowing the oxide ion migration within the oxygen-deficient layer among the sites corresponding to tetrahedral/octahedral units. Here to find out whether the mobile species in La6Sr3Si6O24 are oxygen ions, we conducted tests in different atmospheres of oxygen, nitrogen, and 5% H2 in N2 and found that the electrode responses are almost independent of the atmosphere (Fig. 4a). This phenonium is unlike the typical behavior of electrode response with the partial oxygen pressure change for the oxide ion conductors with good oxide ion conductivities (e.g., > 10−3 S/cm) [50,51]. However, for materials having small and constant carrier concentrations which are determined by intrinsic thermal activation rather than extrinsic oxide-ion substitution from the atmosphere, the oxide-ion conductivity could be independent of pO2 and the inclined electrode response line could be hardly affected by the atmosphere change. Considering low total conductivities within 10−6-10−4 S/cm with large activation energy (~0.93 eV), as well as its atmosphere-independent nature (Fig. 4b) although minor p-type conduction can be still discerned, such low-level ionic conduction in La6Sr3Si6O24 could be still ascribed to the oxygen ions. Such oxide ion conduction could be correlated with the oxygen-vacancy ordered c'-AO2 layers in La6Sr3Si6O24, similar to the hexagonal perovskite oxide ion conductors mentioned above. As indicated by the larger atomic displacement parameter compared with the La2/Sr2 atoms, the La1/Sr1 atoms in the oxygen-deficient c' layers were also considered to be the likely ions for the migration due to their relatively loose packing nature in the structure. However, as indicated by the SEM-EDS results (Fig. S7 in Supporting information), both surfaces of the pellet adjacent to the negative and positive electrodes, and also the cross-section of the pellet parallel to the current direction, show homogeneous elemental distributions and an identical La: Sr: Si ratio with the nominal value (2:1:1) after direct current (DC) polarization. This observation indicated no apparent La or Sr aggregation near the negative electrode side, thus excluding these cations as the ionic conduction species in the sample.
In summary, the 9-layer B-cation and oxygen-deficient shifted hexagonal perovskite-related structure was stabilized on La6Sr3Si6O24 composition under high pressure at 6 GPa. Combined Rietveld refinements of XRD and NPD data indicate B-cation vacancy ordering between two consecutive h-(La/Sr)O3 layers and oxygen vacancy ordering in the c′-(La/Sr)O2 layers, and stacking order of (c′hh)3 in La6Sr3Si6O24. The HP La6Sr3Si6O24 features isolated SiO4 tetrahedral anions between c' and h layers and low-level ionic conduction behavior. The results here indicate that it is practicable to design and synthesize new perovskite-related functional materials from the compositions highly deviated from ABO3 under high pressure.
Declaration of competing interestThe authors declare no competing financial interests.
NotesCSD 2160488 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing datarequest@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: + 44 1223 336033.
AcknowledgmentsThe authors thank the National Science Foundation of China (Nos. 21875049, 22090043 and 22161014), Guangxi Natural Science Foundation (Nos. 2019GXNSFGA245006, AD19245097 and 2020GXNSFAA297220), and the Foundation of Guilin University of Technology (No. GUTQDJJ2018115) for the financial support. We thank Prof. Yonggang Wang (Center for High Pressure Science & Technology Advanced Research, Beijing) for proving facility in the thermal analysis.
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.05.065.
| [1] |
F. Liebau, Structural Chemistry of Silicates. Springer: Berlin, Heidelberg, 1985.
|
| [2] |
D.Y. Pushcharovsky, N.V. Zubkova, I.V. Pekov, Struct. Chem. 27 (2016) 1593-1603. DOI:10.1007/s11224-016-0750-9 |
| [3] |
H. Taniguchi, A. Kuwabara, J. Kim, et al., Angew. Chem. Int. Ed. 52 (2013) 8088-8092. DOI:10.1002/anie.201302188 |
| [4] |
H.P. Wu, B.B. Zhang, H.W. Yu, et al., Angew. Chem. Int. Ed. 59 (2020) 8922-8926. DOI:10.1002/anie.202001855 |
| [5] |
A.J. Fernandez-Carrion, M. Allix, M. Ocana, et al., Inorg. Chem. 52 (2013) 13469-13479. DOI:10.1021/ic401867c |
| [6] |
S. Nakayama, M. Sakamoto, J. Eur. Ceram. Soc. 18 (1998) 1413-1418. DOI:10.1016/S0955-2219(98)00032-6 |
| [7] |
J.G. Wang, S.J. Tian, G.B. Li, et al., Mater. Res. Bull. 36 (2001) 1855-1861. DOI:10.1016/S0025-5408(01)00664-X |
| [8] |
J. Felsche, J. Less-Common Met. 21 (1970) 1-14. DOI:10.1016/0022-5088(70)90159-1 |
| [9] |
F. Nishi, Acta Crystallogr. C 53 (1997) 534-536. |
| [10] |
F.J. Trojer, Z. Kristallogr. 127 (1968) 291-308. DOI:10.1524/zkri.1968.127.1-4.291 |
| [11] |
R.M. Douglass, Am. Mineral. 43 (1958) 517-536. |
| [12] |
A.E. Lapshin, N.B. Borisova, V.M. Ushakov, et al., Russ. J. Inorg. Chem. 51 (2006) 438-444. DOI:10.1134/S0036023606030168 |
| [13] |
A.S. Bhalla, R.Y. Guo, R. Roy, Mater. Res. Innov. 4 (2000) 3-26. DOI:10.1007/s100190000062 |
| [14] |
W.S. Mcdonald, D.W.J. Cruickshank, Acta Cryst. 22 (1967) 37. DOI:10.1107/S0365110X67000076 |
| [15] |
A.K. Pant, D.W.J. Cruickshank, Acta Crystallogr. B 24 (1968) 13. DOI:10.1107/S0567740868001640 |
| [16] |
M. Jansen, R. Hoppe, Z. Anorg. Allg. Chem. 399 (1973) 163-169. DOI:10.1002/zaac.19733990204 |
| [17] |
C.N.W. Darlington, K.S. Knight, Acta Crystallogr. 55 (1999) 24-30. DOI:10.1107/S010876819800963X |
| [18] |
N. Morimoto, K. Koto, Z. Kristallogr. 129 (1969) 65-83. DOI:10.1524/zkri.1969.129.1-4.65 |
| [19] |
T. Yamanaka, Y. Komatsu, M. Sugahara, et al., Am. Mineral. 90 (2005) 1301-1307. DOI:10.2138/am.2005.1621 |
| [20] |
H. Horiuchi, M. Hirano, E. Ito, et al., Am. Mineral. 67 (1982) 788-793. |
| [21] |
Y.B. Wang, F. Guyot, A. Yeganehhaeri, et al., Science 248 (1990) 468-471. DOI:10.1126/science.248.4954.468 |
| [22] |
T. Tsuchiya, J. Tsuchiya, K. Umemoto, et al., Earth Planet. Sci. Lett. 224 (2004) 241-248. DOI:10.1016/j.epsl.2004.05.017 |
| [23] |
M. Murakami, K. Hirose, K. Kawamura, et al., Science 304 (2004) 855-858. DOI:10.1126/science.1095932 |
| [24] |
S.H. Shim, T.S. Duffy, G.Y. Shen, J. Geophys. Res. Solid Earth 105 (2000) 25955-25968. DOI:10.1029/2000JB900183 |
| [25] |
M. Akaogi, H. Kojitani, H. Yusa, et al., Phys. Chem. Miner. 32 (2005) 603-613. DOI:10.1007/s00269-005-0034-1 |
| [26] |
W.S. Xiao, D.Y. Tan, W. Zhou, et al., Am. Mineral. 98 (2013) 2096-2104. DOI:10.2138/am.2013.4470 |
| [27] |
A.A. Belik, W. Yi, J. Phys. Condens. Matter 26 (2014) 163201. DOI:10.1088/0953-8984/26/16/163201 |
| [28] |
K. Fukuda, T. Iwata, E. Champion, Powder Diffr. 21 (2006) 300-303. DOI:10.1154/1.2383066 |
| [29] |
L. Leon-Reina, E.R. Losilla, M. Martinez-Lara, et al., J. Mater. Chem. 14 (2004) 1142-1149. DOI:10.1039/B315257J |
| [30] |
B. Li, W. Liu, W. Pan, J. Power Sources 195 (2010) 2196-2201. DOI:10.1016/j.jpowsour.2009.10.088 |
| [31] |
H. Muller-Bunz, T. Schleid, Z. Anorg. Allg. Chem. 626 (2000) 2549-2556. DOI:10.1002/1521-3749(200012)626:12<2549::AID-ZAAC2549>3.0.CO;2-4 |
| [32] |
H. Muller-Bunz, T. Schleid, Z. Anorg. Allg. Chem. 628 (2002) 564-569. DOI:10.1002/1521-3749(200203)628:3<564::AID-ZAAC564>3.0.CO;2-T |
| [33] |
P.R.Slater Jones, M.S. Islam, Chem. Mater. 20 (2008) 5055-5060. DOI:10.1021/cm801101j |
| [34] |
Y. Masubuchi, M. Higuchi, T. Takeda, Solid State Ionics 177 (2006) 263-268. DOI:10.1016/j.ssi.2005.09.015 |
| [35] |
X.Y. Liu, M.E. Fleet, J. Phys.-Condens. Matter 14 (2002) 11223-11226. DOI:10.1088/0953-8984/14/44/457 |
| [36] |
M.E. Fleet, X.Y. Liu, Am. Mineral. 89 (2004) 396-404. DOI:10.2138/am-2004-2-321 |
| [37] |
J.A. Rodgers, A.J. Williams, J.P. Attfield, Z. Naturforsch. B 61 (2006) 1515-1526. DOI:10.1515/znb-2006-1208 |
| [38] |
M.Camalli Altomare, C. Cuocci, et al., J. Appl. Crystallogr. 42 (2009) 1197-1202. DOI:10.1107/S0021889809042915 |
| [39] |
C.Cuocci Altomare, C. Giacovazzo, et al., J. Appl. Crystallogr. 46 (2013) 1231-1235. DOI:10.1107/S0021889813013113 |
| [40] |
D. Urushihara, T. Asaka, K. Fukuda, et al., Inorg. Chem. 56 (2017) 13007-13013. DOI:10.1021/acs.inorgchem.7b01755 |
| [41] |
R.D. Shannon, Acta Crystallogr. A 32 (1976) 751-767. DOI:10.1107/S0567739476001551 |
| [42] |
S. Zhao, J.J. Yang, Y.F. Han, et al., Chin. Chem. Lett. 34 (2023) 107355. DOI:10.1016/j.cclet.2022.03.078 |
| [43] |
Y.F. Han, Y.J. Zeng, M. Hendrickx, et al., J. Am. Chem. Soc. 142 (2020) 7168-7178. DOI:10.1021/jacs.0c01814 |
| [44] |
F.D. Morrison, D.C. Sinclair, A.R. West, J. Am. Ceram. Soc. 84 (2001) 531-538. DOI:10.1111/j.1151-2916.2001.tb00694.x |
| [45] |
J. Irvine, D.C. Sinclair, A.R. West, Adv. Mater. 2 (1990) 132-138. DOI:10.1002/adma.19900020304 |
| [46] |
S. Fop, J. Skakle, A.C. Mclaughlin, et al., J. Am. Chem. Soc. 138 (2016) 16764-16769. DOI:10.1021/jacs.6b10730 |
| [47] |
S. Fop, K.S. Mccombie, E.J. Wildman, et al., Nat. Mater. 19 (2020) 752-757. DOI:10.1038/s41563-020-0629-4 |
| [48] |
M. Yashima, T. Tsujiguchi, Y. Sakuda, et al., Nat. Commun. 12 (2021) 556. DOI:10.1038/s41467-020-20859-w |
| [49] |
X.J. Kuang, M. Allix, R.M. Ibberson, et al., Chem. Mater. 19 (2007) 2884-2893. DOI:10.1021/cm0626740 |
| [50] |
M. Li, H. Zhang, S.N. Cook, et al., Chem. Mater. 27 (2015) 629-634. DOI:10.1021/cm504475k |
| [51] |
X.Y. Yang, A.J. Fernández-carrióN, J.H. Wang, et al., Nat. Commun. 9 (2018) 4484. DOI:10.1038/s41467-018-06911-w |