 2023, Vol. 34
2023, Vol. 34 


Design of efficient dual-phase emission (DPE) molecules that can exhibit high emission efficiency both in solution and the solid state is of importance for the development of next-generation advanced optical and biomedical materials [1-5]. Generally, aggregation caused quenching (ACQ) and aggregation-induced emission (AIE) are perhaps the most documented phenomena for luminescent molecules [6-11]. ACQ molecules show strong emission in solution but lose luminescence in the solid state, whereas AIE molecules are just the opposite [11-17]. To date, organic luminophore with high emission efficiency both in solution and the solid state is rare because there exists a large structural gap between AIE and ACQ emitters. Therefore, it is still a great challenge for rationally designing and obtaining DPE luminophores.
Organoboron luminophores have long been the flagship of emissive luminophore complexes due to their outstanding emission properties such as high emission quantum yield in solution and tunable emission from visible to near-infrared light [7, 16-23]. However, organoboron luminophores normally show very weak emission in solid state due to ACQ, limiting the practical applications in complex cell microenvironments [3, 14]. Thus, researchers have tried different structural methods to improve their emission efficiency both in solution and in solid state. For example, Niu and co-workers reported a classic organoboron luminophore with a good absolute photoluminescence quantum yield (QY) (ΦF in solution = 85%, ΦF in solid state = 33%) by the adjustment of meso–position group [24]. Qi et al. applied the AIE strategy to obtain a D−A−D dioxaborine derivative showing moderate emission in solution (ΦF in solution = 20.4%) and slightly enhanced one in the solid state (ΦF in solid state = 54.5%) [25]. These studies have successfully constructed organoboron luminophore showing DPE characteristics, whereas leaving plenty of potential for improvement.
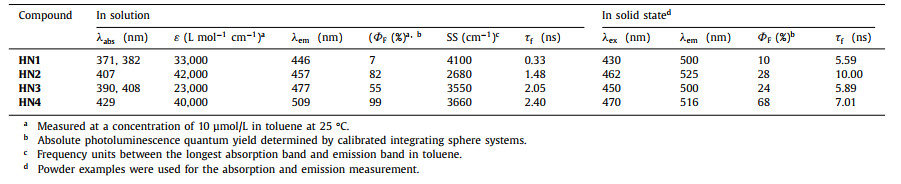
It is known that the emission of traditional organoboron luminophores is quenched in the solid state due to the rigid structure causing deleterious π-π interactions and the self-absorption resulting from very narrow Stokes shifts [26]. Inspired by relevant findings where the gap between rigid structure and self-absorption can be possibly eliminated by structural engineering, herein, we present a rational strategy to create organoboron luminophores for achieving extremely strong DPE. It is expected that based on minimizing the nonradiative transition with a rigid moiety in solution, the twisted core structure can effectively inhibit deleterious π-π interactions in aggregation state. Simultaneously, the emission efficiency of the luminophore can be further enhanced by a moderate intramolecular charge transfer (ICT) effect [27, 28]. In this way, HN1–4 were synthesized via a two or three-step procedure shown in Scheme S1 (Supporting information). The three-ring–fused twisted skeleton was constructed via the boronic coordination of two penta–fluorobenzene and benzothiazole–enolate derived ligands. The carbazole group as an electron donor part was harnessed to the boronic core to form D–A type molecules. Here, HN4 was expected to display unprecedented DPE properties due to an optimization of the twisted core structure and ICT effect. X-ray single-crystal analysis indicated that HN4 had a twisted boronic core structure, while the density functions theory (DFT) calculation showed that its HOMO was mainly localized on the carbazole group, and its LUMO was mainly localized on the boronic core. This supports the ICT nature of HN4.
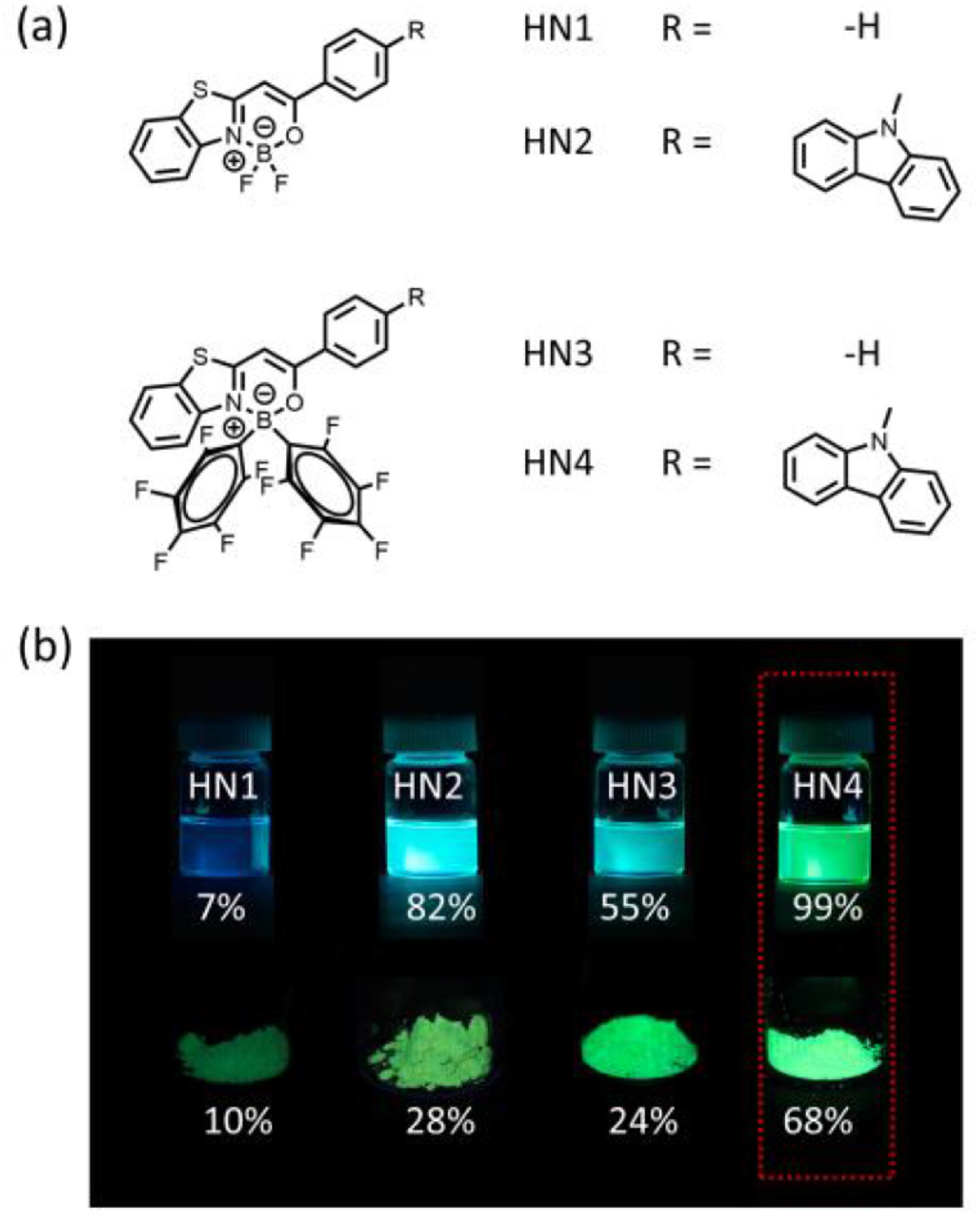
The photophysical properties of HN1–4 were measured in solvents with a different polarity and the solid state. The full details can be found in Fig. 2, Table 1, Figs. S1, S2 and Table S1 (Supporting information). In toluene, HN1 and HN3 showed an absorption maxima at 382 nm (ε = 33, 000 L mol–1 cm–1) and 408 nm (ε = 23, 000 L mol–1 cm–1), respectively. Compared with HN1 and HN3, HN2 and HN4 both showed red shifted absorption bands (~20 nm) with an absorption maxima at 407 nm (ε = 42, 000 L mol–1 cm–1) and 429 nm (ε = 40, 000 L mol–1 cm–1), respectively. Such a red shift of the absorption bands can be explained by ICT enhancement caused by the introduction of carbazole group [26-28]. Besides, compared with HN1 and HN2, HN3 and HN4 also exhibited a red shifted (~20 nm) absorption, respectively, suggesting that two penta–fluorobenzene moieties contribute to the ICT effect as well. Besides, due to the weak absorption characteristic of penta-fluorophenyl-boron, HN3 and HN4 have a lower molar extinction coefficient than HN1 and HN2 [27].

|
Download:
|
| Fig. 1. (a) Chemical structures of compounds HN1–4. (b) Luminescent photograph and QY of HN1–4 in solution and the solid state. | |

|
Download:
|
| Fig. 2. (a) Absorption and (b) normalized emission spectra of HN1–4 (10 µmol/L) in toluene at 298 K. | |
|
|
Table 1 Spectral data of HN1- HN4 in solution and solid state. |
{kind=link}
{kind=link}
From Fig. S1, it is found that the absorption bands of HN1 and HN3 were hardly affected by solvent polarity, while HN2 and HN4 exhibit somewhat solvatochromic effect due to ICT. The maximum emission wavelength (Fmax) of HN1 and HN3 was at 446 nm and 477 nm, respectively, showing almost no wavelength variation with the change of solvent polarity (Fig. S2). However, their emission intensity was affected by the solvent polarity, indicating that there existed a weak ICT in HN1 and HN3. In contrast, Fmax and emission intensity of HN2 and HN4 were significantly affected by solvent polarity, and two shoulder peaks were observed in low polar solvents due to the mirror image of the absorption spectrum. They showed a dramatically red-shifted emission in high polar solvents such as dimethyl sulfoxide (DMSO), suggesting that the ICT effect played a significant role in the photophysical properties of these molecules [1].
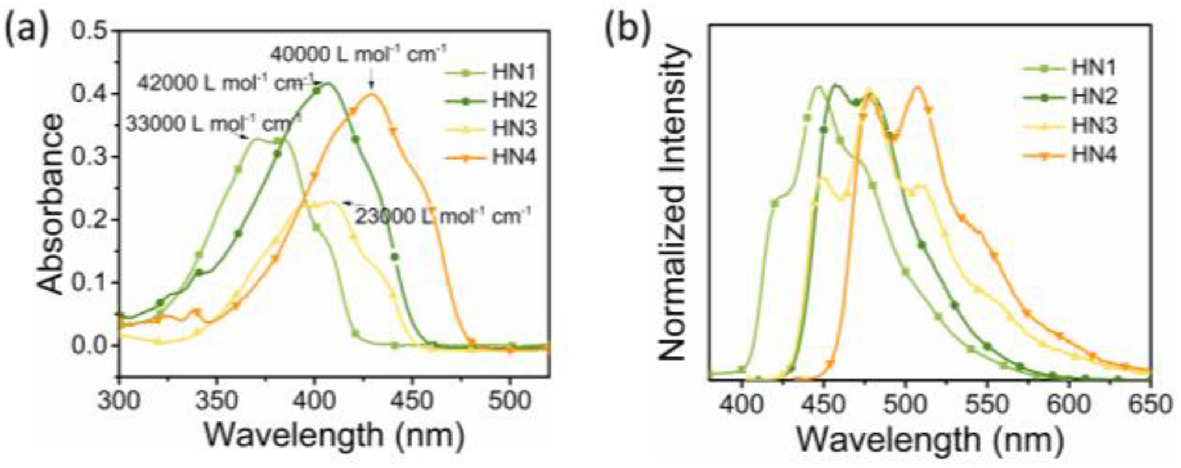
Due to electronic donor effect of carbazole, HN2 and HN4 showed strong luminescence in solution (Fig. 3, Figs. S2 and S3 in Supporting information). Notably, as shown in Figs. 3a and b, HN4 expressed outstanding QY in any organic solvent (e.g., ΦF in solution = 88% in hexane, 99% in toluene, 92% in CHCl3, 33% in DMSO which remained high), suggesting that moderate ICT effect can further enhance the radiative process along with an increased Stokes shift, whereas strong ICT, on the contrary, is a quenching factor due to an enhancement in the rate of nonradiative decay (Table S1) [27, 28]. HN4 showed the highest QY in toluene due to the effect between aromatic molecules and toluene [3]. It is emphasized that the luminescent color of HN2 changed from blue to yellow with increased solvent polarity (Fig. S3), while HN4 showed light green in low-polarity solvents and changed to yellow in the high-polarity solvents (Fig. 3a) because of ICT.

|
Download:
|
| Fig. 3. (a) Photograph under 365 nm UV lamp and (b) emission spectra of HN4 (10 µmol/L) in hexane, toluene, CHCl3 and DMSO at 298 K, λex = 429 nm. (c, d) Emission spectra (λex = 429 nm) and (e) photograph under 365 nm UV lamp of HN4 in THF–water mixtures (10 µmol/L) at 298 K with varying volumetric fractions of water (fw). | |
{kind=link}
In the solid state, HN1–4 showed a red shifted (about 50 nm) absorption maxima at 430 nm, 462 nm, 450 nm and 470 nm, respectively, compared with those in toluene solution (Table 1). The emission bands of HN1–4 remained narrow and the Fmax values (500–525 nm) also underwent a red shift relative to those in toluene solution (Table 1 and Fig. S5 in Supporting information). Such a shift is due to the aggregation–induced CT enhancement [2, 7]. While referring to the emission shift, it should be noted that the Fmax values of HN1 and HN2 showed a red shift of more than 50 nm, whereas HN3 and HN4 showed a red shift less than 50 nm. These results suggest that twisted structure based on two penta-fluorobenzenes had effectively weakened the deleterious intermolecular π-π interactions in the solid state by increasing the intermolecular spacing, as close molecular stacking could readily cause a spectral red-shift [14]. Especially, HN4 only showed a red shift of 7 nm of its emission in the solid state compared to that in solution, indicating that the π-π interactions for HN4 are suppressed to a huge degree in the solid state (Table 1).
As expected, HN1 and HN3 showed moderate QY in toluene solution (ΦF in solution = 7% − 55%), whereas HN2 and HN4 showed outstanding QY (ΦF in solution = 82% − 99%) (Table 1). Importantly, HN2–4 also showed intense emission in the solid state with a high QY (ΦF in solid state = 28%, 24% and 68%), respectively. Besides, it can be found that only the QY increased significantly upon the introduction of penta–fluorobenzene. The photoluminescence lifetime among HN1–4 remained at nanosecond scale both in solution and in the solid state. This indicates that the possible isomers in carbazole group have little effect on our study [29]. To the best of our knowledge, HN4 showed almost the highest dual-phase QY among organoboron luminophores so far. Besides, the photoluminescent lifetime of HN4 is about 8 times higher than that of HN1 in solution (Table 1 and Fig. S6 in Supporting information). These results suggest that our strategy of creating high-efficient DPE organoboron luminophore by the unique structural engineering, rigid-twisted core structure with moderate ICT effect, is successful.
To investigate the possible further tunability of QY, the emission spectra of HN1–4 were investigated in THF/H2O mixtures with various ratios to compare the photophysical properties of these molecules in different aggregated states (Figs. 3c–e, Fig. S4 in Supporting information). The emission intensity of HN1 and HN3 remained almost the same until the water fraction (fw) reached 80%. Upon addition of 90% water into THF, HN1 showed large red–shifted emission from 442 to 492 nm and the emission color changed from blue to green, while HN3 remained the initial emission wavelength and the emission color. On the other hand, HN2 and HN4 showed a gradual decrement of the emission intensity with the addition of water into the THF solution till 70% and 60% of fw, respectively. Further increase of fw made their emission intensity enhanced again. Especially, HN4 still showed appreciable luminescence QY (28%), even when the emission intensity turned to be the lowest in fw of 60%. These results should be largely due to the twisted skeleton constructed via the boronic coordination of two penta-fluorobenzene.
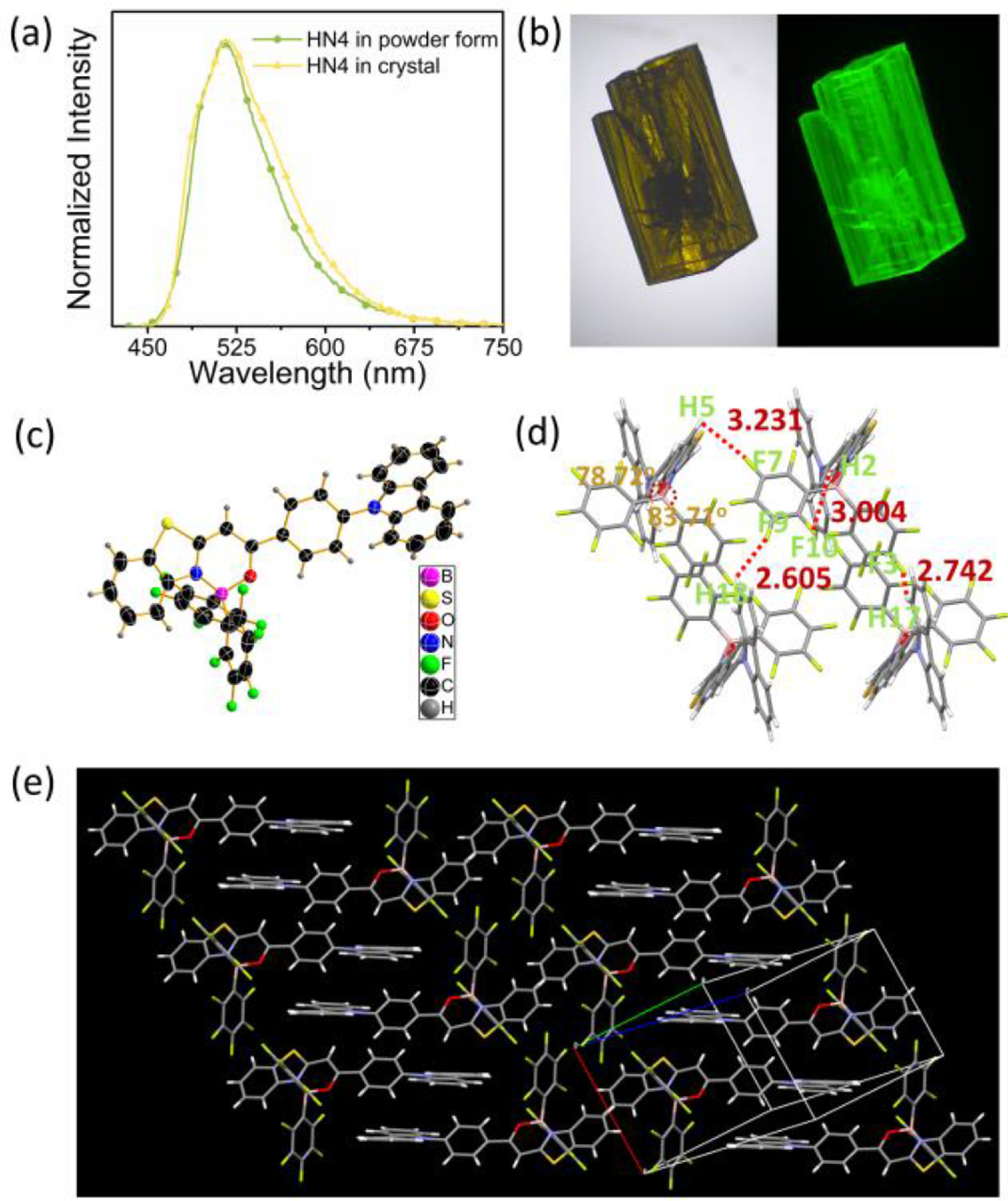
In order to better understand the molecular mechanism for these strong emissions, X-ray crystallographic analysis was performed, since there are the same Fmax values of HN4 in the solid states as powder form or as crystal form (Fig. 4a). The crystal of HN4 showed yellow color in bright field and a bright green luminescence under 450 nm laser light in Fig. 4b. The ORTEP (Oak Ridge Thermal Ellipsoid Plot) drawing and molecular packing structure of HN4 are shown in Fig. 4 and Fig. S10 (Supporting information). The boron atoms adopt a typical tetrahedral geometry to form N^O–chelate six-membered ring, which contributes to the construction of the rigid three-ring-fused π-conjugated skeletons. There have been reports showing that intermolecular spacing can be effectively increased after introducing phenyl into boron atom [27, 33]. As anticipated, HN4 adopts rigid–twisted core conformation, making the molecular stacking of HN4 J-aggregation (Fig. 4e). The six-membered ring of the boron core is not on a plane because of the steric hindrance of penta-fluorobenzenes, and the dihedral angle is 24.90°. The dihedral angles of the N-B-O plane and two penta–fluorobenzenes are 78.72° and 83.71°, respectively. The plane of carbazole forms an obvious angle (47.96°) with the N-B-O plane. Intermolecular π-π interactions were not detected in HN4 upon molecular packing mode. However, multiple short interatomic contacts existed within the crystals: F7···H5–C5 (80.17°, 3.231 Å), F9···H18–C18 (125.34°, 2.605 Å), F10···H2–C2 (154.52°, 3.004 Å), F3···H17–C17 (157.15°, 2.742 Å). Furthermore, C–H···π interactions were detected on 2.869 Å–3.019 Å. These weak intermolecular interactions inhibit the molecular internal rotations and block the non–radiative relaxation, and the twisted skeleton constructed effectively inhibited the π-π interaction, causing HN4 to show intense emission in the solid state.

|
Download:
|
| Fig. 4. (a) Emission spectra of HN4 in the powder and crystal form, λex = 470 nm. (b) Crystal photograph of HN4 in bright field (left) and under 450 nm laser light (right). (c) Molecular structures of HN4 (50% probability for thermal ellipsoids). (d) Crystal packing structure of HN4. The red dotted lines show intermolecular interactions. (e) Crystal packing structures of HN4 from a monomolecular layer view. | |
{kind=link}
To further illustrate the effect of the structure on the electronic structures which are related to the photophysical properties of HN1–4, DFT calculation was carried out [27]. As shown in Fig. S7 (Supporting information), HN1 and HN3 have HOMOs and LUMOs localized over the whole molecules. The calculated first excited state, mainly consisting of HOMO→LUMO transition, has excitation energy of 3.47 eV (358 nm, f = 0.8577) for HN1 and 3.32 eV (374 nm, f = 0.5863) for HN3, respectively. The incorporation of the penta-fluorobenzene group in HN3 led to an increase of the HOMO and LUMO energy level; however, the HOMO–LUMO gap was decreased. Meanwhile, the incorporation of carbazole group in HN2 is predicted to greatly increase the HOMO energy level by 0.62 eV, but to have little effect on the LUMO, thus decreasing the HOMO–LUMO gap and resulting in a red-shift of the main absorption band relative to those of HN1. The energy levels of HOMOs of HN2 (3.05 eV, 406 nm, f = 0.5749) and HN4 (3.00 eV, 414 nm, f = 0.4732) are increased relative to those of HN1 and HN3, due to the introduction of the electron-donating carbazole. The decreased HOMO-LUMO gaps of HN2 and HN4 should be responsible for the red-shifted main absorption band relative to those of HN1 and HN3. HN2 and HN4 have HOMOs dominated by phenyl-carbazole group, whereas their LUMOs are mostly localized on the benzothiazole and phenyl unit. It is obvious that the lowest-energy excited state for HN2 and HN4 corresponds to a charge transfer from the carbazole fragment to the benzothiazole unit. The features of charge transfer make the ground and excited states more energetically distinct, leading to large Stokes shifts and a reduction in self-absorption.
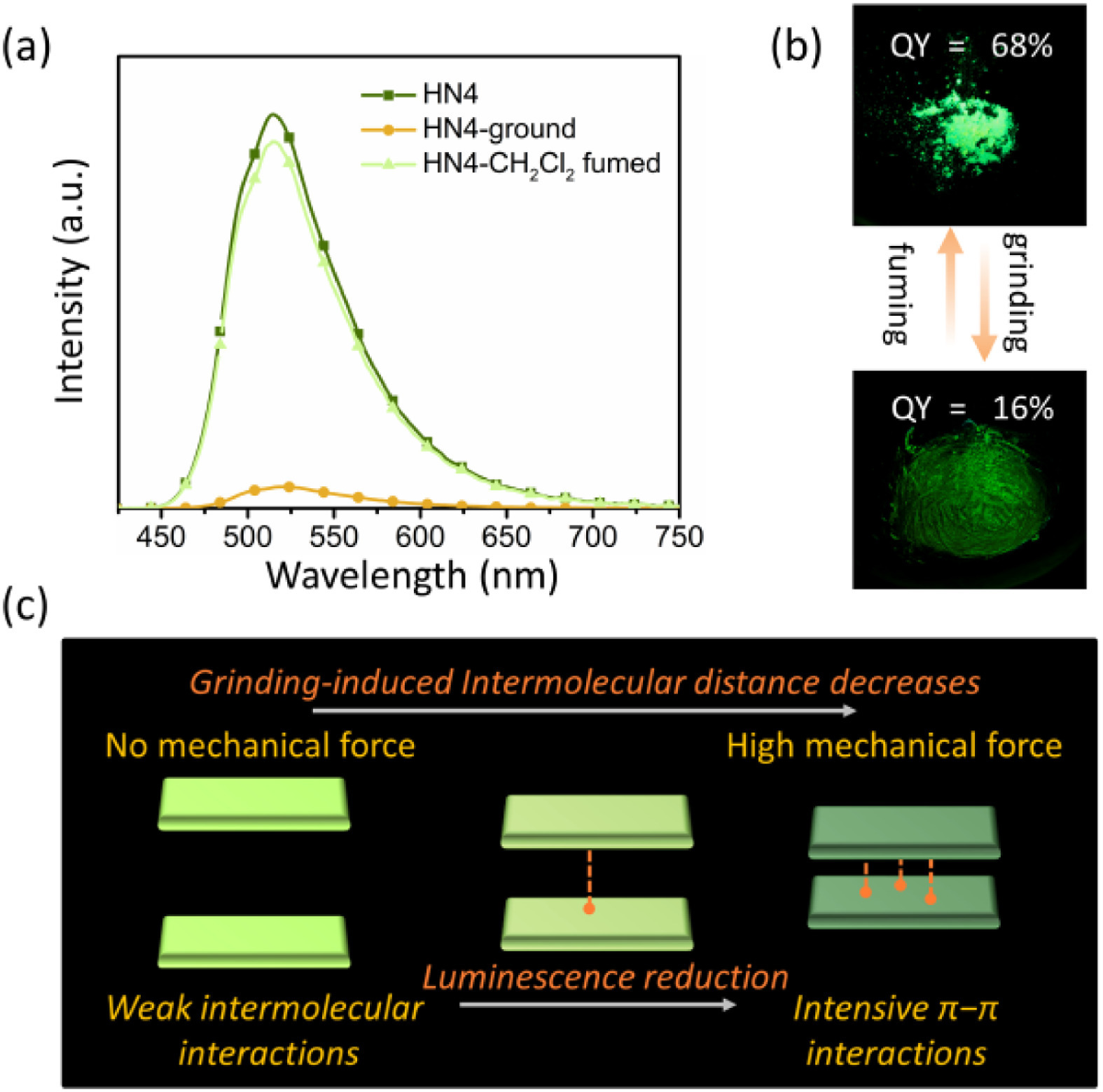
Considering external mechanical force that can often cause structural reorganization in the solid state, resulting in a change of material luminescence signal [30-32], mechanical stimuli responsive properties for HN4 were also studied (Fig. 5). As expected, the QY of HN4 was significantly reduced upon grinding with a pestle for 5 min. Its original luminescence of the powder sample was gradually recovered when treated with CH2Cl2 vapor for a few minutes. In addition, the grinding led to distinct changes in their powder X-ray diffraction (XRD) patterns (Fig. S8 in Supporting information). All the diffraction peaks of HN4 were weakened due to the higher amorphization of molecular packing, originating from the decrease of intermolecular distance that resulted in the deleterious π-π interactions and luminescence reduction [32]. However, HN4 still showed moderate luminescence QY (16%) after grinding, and only the QY difference relative to that of the initial solid state is huge. This perfect reversibility also indicated that such a mechanical grinding is a physical process to change the molecular interaction, rather than any chemical reaction or oxidation effect. This mechanical stimuli-responsive reversible QY alternation endowed the solid state material with superior molecular switch performance.

|
Download:
|
| Fig. 5. (a) Emission spectra of HN4 in the solid state initially, after being ground, and then followed by CH2Cl2 fuming, λex = 470 nm. (b) Photographs of the powder of HN4 before and after grinding under UV 365 nm lamp. (c) Proposed mechanism of the molecular switch performance for HN4. | |
{kind=link}
In summary, we have developed a practical molecular engineering strategy by constructing a rigid-twisted core structure and moderate ICT effect to obtain outstanding DPE organoboron luminophores. The rigid π-conjugated skeletons can enable radiative energy dissipation, allowing luminophore with high emission efficiency in solution, and the twisted core structure of these compounds could enhance the emission efficiency in the solid state by significantly reducing the deleterious intermolecular π-π stacking. Concurrently, the emission efficiency of the luminophore can be further enhanced by a moderate ICT effect. Importantly, HN4 showed almost the highest dual-phase QY among organoboron luminophores so far. Furthermore, HN4 is capable of the mechanical force sensibility by the reversible change of QY, which may potentially serve as a superior solid state molecular switch performance. We believe that this could be a valuable strategy to design DPE materials to benefit their practical applications in various luminescent events.
Declaration of competing interestWe declare that we have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 21975046) and partially from the National Key Research and Development Program of China (No. 2017YFA0207700).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.06.035.
| [1] |
J.L. Belmonte-Vázquez, Y.A. Amador-Sánchez, L.A. Rodríguez-Cortés, et al., Chem. Mater. 33 (2021) 7160-7184. DOI:10.1021/acs.chemmater.1c02460 |
| [2] |
T. Zhang, X. Ma, H. Tian, Chem. Sci. 11 (2020) 482-487. DOI:10.1039/C9SC05502A |
| [3] |
G. Chen, W. Li, T. Zhou, et al., Adv. Mater. 27 (2015) 4496-4501. DOI:10.1002/adma.201501981 |
| [4] |
H. Wu, Z. Chen, W. Chi, et al., Angew. Chem. Int. Ed. 58 (2019) 11419-11423. DOI:10.1002/anie.201906507 |
| [5] |
S. Feng, Z. Qu, Z. Zhou, et al., Chem. Commun. 57 (2021) 11689-11692. DOI:10.1039/D1CC04687J |
| [6] |
J. Luo, Z. Xie, L. Cheng, et al., Chem. Commun. 18 (2001) 1740. |
| [7] |
H. Lu, J. Mack, Y. Yang, et al., Chem. Soc. Rev. 43 (2014) 4778-4823. DOI:10.1039/C4CS00030G |
| [8] |
Y. Xie, Z. Li, Natl. Sci. Rev. 8 (2021) nwaa199. DOI:10.1093/nsr/nwaa199 |
| [9] |
X. Jia, C. Shao, X. Bai, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 4816-4821. DOI:10.1073/pnas.1821991116 |
| [10] |
Y. Zhou, J.W. Kim, R. Nandhakumar, et al., Chem. Commun. 46 (2010) 6512-6514. DOI:10.1039/c0cc01715a |
| [11] |
V. Nguyen, Y. Yim, S. Kim, et al., Angew. Chem. Int. Ed. 59 (2020) 8957-8962. DOI:10.1002/anie.202002843 |
| [12] |
E. Galbraith, T.D. James, Chem. Soc. Rev. 39 (2010) 3831-3842. DOI:10.1039/b926165f |
| [13] |
Z. Guo, S. Park, J. Yoon, et al., Chem. Soc. Rev. 43 (2014) 16-29. DOI:10.1039/C3CS60271K |
| [14] |
K. Li, X. Duan, Z. Jiang, et al., Nat. Commun. 12 (2021) 2376. DOI:10.1038/s41467-021-22686-z |
| [15] |
H. Dolati, L.C. Haufe, L. Denker, et al., Chem. Eur. J. 26 (2020) 1422-1428. DOI:10.1002/chem.201905344 |
| [16] |
J. Chen, W. Liu, X. Fang, et al., Chin. Chem. Lett. 33 (2022) 5042-5046. DOI:10.1016/j.cclet.2022.03.120 |
| [17] |
Y. Zhang, L. She, Z. Xu, et al., Chin. Chem. Lett. 33 (2022) 3277-3280. DOI:10.1016/j.cclet.2021.11.004 |
| [18] |
M.K. Kuimova, G. Yahioglu, J.A. Levitt, et al., J. Am. Chem. Soc. 130 (2008) 6672-6673. DOI:10.1021/ja800570d |
| [19] |
S. Erbas-Cakmak, S. Kolemen, A.C. Sedgwick, et al., Chem. Soc. Rev. 47 (2018) 2228-2248. DOI:10.1039/C7CS00491E |
| [20] |
Y. Zhou, Y. Zhuang, X. Li, et al., Chem. Eur. J. 23 (2017) 7642-7647. DOI:10.1002/chem.201700947 |
| [21] |
H. Li, F. Lv, X. Guo, et al., Chem. Commun. 57 (2021) 1647-1650. DOI:10.1039/D0CC07961H |
| [22] |
M. Li, S. Long, Y. Kang, et al., J. Am. Chem. Soc. 140 (2018) 15820-15826. DOI:10.1021/jacs.8b09117 |
| [23] |
X. Ma, J. Wang, H. Tian, Acc. Chem. Res. 52 (2019) 738-748. DOI:10.1021/acs.accounts.8b00620 |
| [24] |
C. Duan, Y. Zhou, G.G. Shan, et al., J. Mater. Chem. C 7 (2019) 3471-3478. DOI:10.1039/C8TC06421K |
| [25] |
Y. Qi, Y. Wang, G. Ge, et al., J. Mater. Chem. C 5 (2017) 11030-11038. DOI:10.1039/C7TC02115A |
| [26] |
Q. Liu, X. Wang, H. Yan, et al., J. Mater. Chem. C 3 (2015) 2953-2959. DOI:10.1039/C4TC02876G |
| [27] |
Y. Wu, H. Lu, S. Wang, et al., J. Mater. Chem. C 3 (2015) 12281-12289. DOI:10.1039/C5TC03084F |
| [28] |
N. Venkatramaiah, G.D. Kumar, Y. Chandrasekaran, et al., ACS Appl. Mater. Inter. 10 (2018) 3838-3847. DOI:10.1021/acsami.7b11025 |
| [29] |
C. Chen, Z. Chi, K.C. Chong, et al., Nat. Mater. 20 (2021) 175-180. DOI:10.1038/s41563-020-0797-2 |
| [30] |
H. Yu, W. Ren, H. Lu, et al., Chem. Commun. 52 (2016) 7387-7389. DOI:10.1039/C6CC02937J |
| [31] |
Y. Liu, Q. Zeng, B. Zou, et al., Angew. Chem. Int. Ed. 57 (2018) 15670-15674. DOI:10.1002/anie.201810149 |
| [32] |
X. Wang, Q. Liu, H. Yan, et al., Chem. Commun. 51 (2015) 7497-7500. DOI:10.1039/C5CC01902H |
| [33] |
Y. Wu, Z. Li, Q. Liu, et al., Org. Biomol. Chem. 13 (2015) 5775-5782. DOI:10.1039/C5OB00607D |