 2022, Vol. 33
2022, Vol. 33 


b Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Suzhou 215006, China;
c Laboratory of Ion Beam Physics, ETH Zurich, Zurich 8093, Switzerland;
d Baotou Research Institute of Rare Earths, Baotou 014030, China
Plutonium-244 (244Pu), with a half-life of 81.1 ± 0.3 Myr [1], could only be produced in the rapid neutron-capture process (r-process) under the high neutron density and high-temperature stellar environment. 244Pu could be the heaviest and the shortest-lived pure r-process primordial nuclide that may still remain detectable on Earth [2]. There is natural and anthropogenic 244Pu on Earth, and the natural 244Pu is produced only through r-process and can be classified into primordial and live interstellar. The primordial 244Pu is supposed to be produced in the stellar evolution during the formation of the Earth, and the existence of which has been indirectly proven by precise measurement of excessive 244Pu-fissiogenic xenon isotopes found in achondritic meteorites [3]. The initial abundance ratio of 244Pu to 238U for early Solar System ((244Pu/238U)ESS) was estimated to be ~0.008 from Xe isotopic loss in ancient zircon samples [4]. In the past millions of years, the interstellar medium (ISM, composing of diffused gas clouds and small solid particles) ejected by supernova explosion near the solar system could contain the live interstellar 244Pu, which may eventually be deposited on Earth and buried in ocean floor with the ejected ISM dust particles [5]. In addition, anthropogenic 244Pu can be produced in high flux reactors and thermonuclear detonation events, and it was only found at very low levels (with a 244Pu/239Pu ratio of approximately (5.7 ± 1.0) × 10−5 in the top layers of surface soil and sediment samples [6].
In recent years, the flux and accumulation rate of live interstellar 244Pu in sediment and deep-sea iron-manganese encrustation were analyzed to estimate the frequency of r-process and to explain the mechanism of stellar evolution [5, 7, 8]. Due to very similar chemical properties between actinides and lanthanides, primordial 244Pu is most likely to be enriched and thus found in rare earth minerals. Several attempts have been made to search for primordial 244Pu in rare earth samples, including bastnaesite and gadolinite samples, for verification of its abundance produced in the formation of the solar system [9-11], especially its ratio to heavy nuclides, which is an important parameter for validating the theories and models of galactic nucleosynthesis, chronology of solar system and terrestrial evolution. In 1971, the content of 244Pu had been first reported to be 1 × 10−18 g/g in a bastnaesite sample obtained from the Mountain Pass Mine (California, USA) by Hoffman and Lawrence [1]. However, subsequent studies for searching such a 244Pu signal, either via indirect method [12] by searching the fission tracks of 244Pu or via direct detection [10] of 244Pu with an upper limit of 10 folds lower than Hoffman's, failed to confirm the concentration of 244Pu reported by Hoffman and Lawrence. It should also be noted that the bastnaesite samples used in the previous direct detection of 244Pu [1, 9, 10] were not completely dissolved and measured, which could lead to under-estimation of the 244Pu results. Thus, in the earlier study, we have developed an ultra-sensitive analytical method for measuring total 244Pu in completely digested bastnaesite samples by accelerator mass spectrometry (AMS) [11]. For complete dissolution of 22.27 g bastnaesite concentrate sample, an upper limit of ~4.5 × 10−19 g/g 244Pu was obtained at 99% confidence level, which still disapproved the detection value reported in the reference [1]. Therefore, a larger bulk sample need to be processed to verify the abundance of primordial 244Pu.
In this work, the abundance of 244Pu in 450 g of bastnaesite concentrate sample from the Bayan Obo ore deposit is measured using the ultra-sensitive AMS method developed previously [11]. The enrichment factor and the abundance of 244Pu are estimated, and the acid leaching behavior of Pu is discussed.
The direct detection of primordial 244Pu in terrestrial samples would be extremely difficult as it should have decayed to an almost immeasurable fraction (1.1 × 10−17) of its initial abundance. Owing to the processes of geochemical enrichment, the abundance of an element in a favourable host mineral can exceed its average terrestrial abundance by a large (enrichment) factor. Primordial 244Pu is most likely to be enriched in rare earth mineral samples because of the similar chemical properties of actinides and lanthanides. The bastnaesite sample used in the present study was derived from the Bayan Obo deposit (China) formed in Mesoproterozoic Era about 1.3 Ga ago. The principle rare earth minerals are bastnaesite (REFCO3) and monazite (REPO4) with the ratio of about 9:1 to 1:1, and the total REO content in the sample was determined to be 49.3% [13]. 239Pu and 240Pu in this bastnaesite concentrate sample was measured to be not above 0.1 fg/g level, confirming that the sample used in the present study has not been contaminated by anthropogenic Pu.
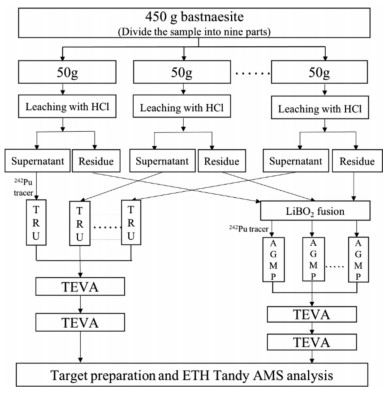
A set of samples, including one 450 g and one 50 g bastnaesite concentrate sample as well as two of procedural blank samples, were analyzed. A flow diagram of the sample preparation procedure is given in Fig. 1. The 450 g sample was divided into nine of ~50 g samples for pretreatment and the first stage column separation, all the eluate samples were then mixed together for the subsequent TEVA resin purification. The details of the column separation and purification method have been described elsewhere [11]. For the sample preparation, the bastnaesite concentrate samples were firstly leached by 12 mol/L HCl, and the supernatant solution and the residue were obtained, respectively. About 70% of the REEs and 9% of the Th dissolved in the HCl leachate, while ~90% of Th and ~30% of REEs remained in the residue fraction. The residue was dried and mixed well with lithium metaborate, lithium iodide and sodium persulfate, and fused at 950 ℃. The fusion button was dissolved in mixed acid (6 mol/L HCl + 4 mol/L HNO3). About 1–2 pg of the 242Pu tracer was added to the HCl leachate and the solution of the fusion button for tracing the chemical recovery, and nine parts of the 450 g sample were added with 200 fg of the 242Pu, and the two solutions were then separately processed. For Pu separation and purification of the HCl leachate, a TRU column was used to directly extract Pu in the HCl leachate, and Pu in the eluate was further purified using the TEVA column repeatedly to remove residual REE and Th. For the separation and purification of the bastnaesite residue solution, NaOH was added to co-precipitate Pu with REE hydroxide. The precipitate was dissolved with HCl, and Pu was separated using an AGMP-1 M anion resin column. The eluate from the AGMP-1 M column was also purified with the TEVA resin repeatedly. The AMS target was then prepared for the TEVA eluate sample using mixed titanium and iron hydroxide co-precipitation method with 0.4 mg of Ti and 0.1 mg of Fe [14]. The Fe/Ti hydroxide precipitate was subsequently baked at 650 ℃ for conversion to the oxide form. The oxide sample was finally mixed with niobium powder and pressed into a Ti target holder for AMS measurement.

|
Download:
|
| Fig. 1. Flow diagram of the sample preparation procedure. | |
The isotopic ratios of the analytes to the tracer (i.e., 239Pu/242Pu and 244Pu/242Pu) in the HCl leachate and the residue of these bastnaesite concentrate samples were measured by the low energy (300 kV) AMS system TANDY at ETH Zurich [15]. This compact AMS was built in late 1990s, and the system had been significantly modified in order to improve its performance, including a new gas ionization detector, an upgraded ion source, a second HE magnet, and the implementation of He stripping [16], which significantly increased the overall transmission for actinides [15]. Also the AMS target preparation method had been effectively optimized, which further improved analytical accuracy and precision [17].
The details of the AMS setup for the measurements of actinides have been described elsewhere [14, 18]. To achieve lower 244Pu detection limits, each sample target has been measured for ~6.5 h and nearly sputtered until exhaustion. Only the contents of 239Pu and 244Pu were analyzed in the present study, and a large portion of the AMS measurement time was allocated to 244Pu (62.5%) to achieve the possibly lowest detection limits for 244Pu. Good and consistent total efficiencies were obtained for the HCl leachates and the residues of the two bastnaesite concentrate samples (450 and 50 g), but the efficiencies for the procedural blank samples were significantly lower, which may be due to a loss of Pu in the column separation. The total efficiencies (chemical yield included) of Pu isotopes in the present study were respectively 2.40 × 10−4 for the HCl leachate and 4.20 × 10−4 for the residue of the 450 g sample, which were ~20 times higher than the achieved total efficiency of 244Pu (2.2 × 10−5) reported by Lachner et al. [10].
Although the main sources of 239Pu in the environment are anthropogenic, small amounts of 239Pu can be produced naturally via neutron capture on 238U [19], and the occurrence of natural 239Pu was subsequently demonstrated in uranium ores by a series of studies [20, 21]. The ratio of 239Pu/238U has been estimated to be (0.1–3) × 10−12 in uranium ore samples [21]. The natural 239Pu content in our bastnaesite samples can thus be calculated to be (0.19–5.7) × 10−18 g/g taking into account the 238U content measured by alpha spectrometry (1.90 × 10−6 g/g). As most of the Th (> 90%) retained in the residue after leaching with concentrated HCl [11], it would be skeptical that the Pu, which is also stable at +4 valence with very similar chemical properties to Th(IV), may not be fully released by acid leaching. In order to verify the necessity of complete dissolution of the sample and measurement of 244Pu in the leaching residue, the two sample fractions (i.e., the HCl leachate and the digested solution of the residue) were processed separately, and the contents of 239Pu were analyzed. The masses of 239Pu were calculated from the measured and blank corrected atomic ratios of 239Pu to the known amounts of 242Pu added as tracer. The 239Pu for the HCl leachates and the residue solutions of the bastnaesite samples are listed in Table 1.
|
|
Table 1 Results for 239Pu in bastnaesite concentrate samples and reagent blanks. |
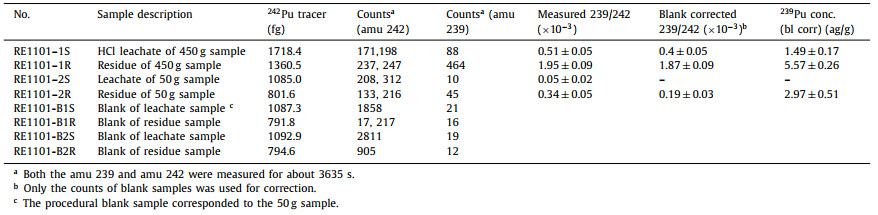
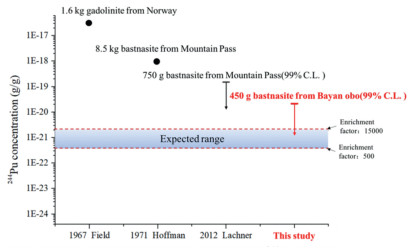
Attogram level of 239Pu were detected in the AMS targets of both bastnaesite samples, and the counting rates of 239Pu in the 450 g sample are ~9 and 10 times higher than those of the 50 g sample, which obviously correlated with the amount of sample or the reagent used. Since the counting rate of 239Pu in the residue sample was ~4.5 times higher than that of the HCl leachate sample, 239Pu may come from two sources. One possibility was that the 239Pu came from the reagents used in the sample preparation procedure (particularly LiBO2 for the fusion of the residue and NaOH for the coprecipitation step). The other source was indeed the presence of natural 239Pu in the bastnaesite sample, implying that majority (> 70%) of the natural Pu may still retained in the residue (similar to Th) after leaching with HCl. Then, in the future studies, the rare earth sample needs be completely dissolved to ensure that the 244Pu content is not under-estimated; meanwhile, it also suggests that the samples containing a high Th content could be more suitable for searching primordial 244Pu due to the very close chemical behaviors of these two elements [11]. Unfortunately, due to the poor chemical recoveries of the procedural blanks, the content of 239Pu in the blank samples could not be accurately measured, and possible contributions of 239Pu from the chemical reagents used in the sample preparation procedure could not be completely excluded. As a consequence, it is not appropriate to use the 239Pu/242Pu ratio of the blanks to correct the samples for the 239Pu contamination because this method assumes that all 239Pu had been introduced together with the 242Pu tracer. Instead, we used the average counting rate of 239Pu from the blank samples for correction. Still, the origin of 239Pu in the sample needs to be further analyzed and verified. Even if 239Pu came from the Global Fallout contamination, since the maximum contamination level 244Pu/239Pu ratio is 1 × 10−5 [22], the maximum level of 244Pu from Global Fallout in our sample were not excess 5 × 10−23 g/g, which is significantly lower than the expected range of primordial 244Pu in the rare earth minerals (Fig. 2).

|
Download:
|
| Fig. 2. Comparison of analytical concentration of 244Pu in rareearth mine. | |
For the 244Pu measurement in bastnaesite concentrate, no 244Pu signal was detectable for all the HCl leachate and the residue samples. Based on the total efficiency of Pu, one count of 244Pu in the 450 g sample would equal to 5.6 × 10−21 g/g and 3.2 × 10−21 g/g for the HCl leachate and the residue, respectively. This means, if there were 14 244Pu atoms for the HCl leachate or 8 244Pu atoms for the residue contained in one gram of bastnaesite concentrate, a 244Pu signal would have been detected. An upper detection limit (UDL) for the 244Pu content in the HCl leachate or the residue of the bastnaesite concentrate can be estimated (Table 1) using the equation below:
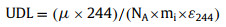
|
where μ is the upper limit counts at 99% C.L., corresponding to 4.74 counts for a sample with zero background and zero count observed [23]; NA is the Avogadro constant (i.e., 6.022 × 1023); mi is the bastnaesite mass processed for individual sample; and ε244 is the total 244Pu efficiency for individual sample (Table 2).
|
|
Table 2 Results for 244Pu in bastnaesite concentrate samples and reagent blanks. |
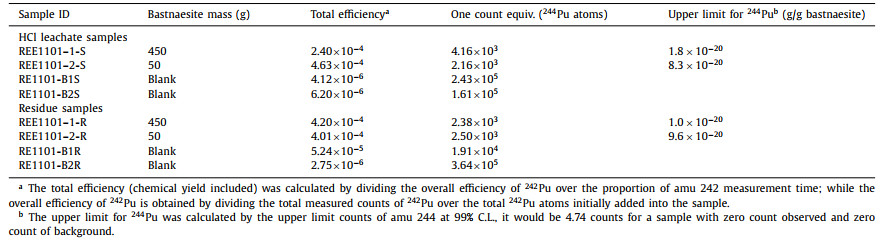
The upper detection limits were calculated to be 1.8 × 10−20 g 244Pu and 1.0 × 10−20 g 244Pu per gram bastnaesite for the HCl leachate and the residue of the 450 g sample at 99% confidence level, respectively. Thus, the upper detection limit of 244Pu in total bastnaesite concentrate at 99% confidence level is 2.1 × 10−20 g/g, calculating by combined upper detection limits of the HCl leachate and the residue (i.e., 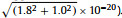
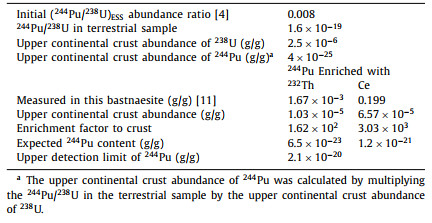
Estimation of 244Pu abundance. The initial (244Pu/238U)ESS abundance ratio in the early formation of the solar system 4.57 billion years ago has been determined to be ~0.008 from the measurements of a fissiogenic xenon excess in meteorites and in 4.1–4.2 billion-year-old zircons. Due to the radioactive decay of the 238U and 244Pu, the existing 244Pu on earth has decayed to a fraction of 1.1 × 10−17 to its initial concentration since the formation of the solar system and 238U reduced by half. Then, the present abundance ratio of primordial 244Pu/238U is expected to be around 1.6 × 10−19 in terrestrial samples. The upper limit of the 244Pu content in the Earth's crust is deduced to be 4 × 10−25 g/g from the upper continental crust abundance of 238U (i.e., 2.5 × 10−6 g/g) [24]. Owing to geochemical enrichment processes, the abundance of 244Pu in a rare earth mineral can exceed its average terrestrial abundance by a large enrichment factor. However, due to the uncertainty of plutonium oxidation state during high-temperature geological history of rare earth ore bodies, it would be difficult to accurately estimate this enrichment factor. Since the most stable oxidation states of Pu are tetravalent and trivalent, plutonium is very likely to be enriched in tetravalent state with thorium(IV) and cerium(IV) in the oxidizing environment, and concentrated with trivalent rare earth elements in reducing environment.
Wedepohl [24] reported that the discrete ratios of fissiogenic xenon components of 244Pu in an ancient igneous rock seemed closely associated with the light REEs. CeO2 has been often used as a surrogate for PuO2 when studying ZrO2−MgO matrix nuclear fuel containing PuO2 [25]. Jones and Burnett [26] reported that Pu and light REE (especially cerium) in meteorite material were almost geochemically identical in reducing environment with very little fractionation. Thus, cerium could be a chemical and structural homologue of plutonium, and their trivalent and tetravalent oxide species could be enriched concurrently in geological environment. Using the similar enrichment factor of Pu to Ce, the upper limit of 244Pu content in this bastnasite sample can be calculated as 1.2 × 10−21 g/g, i.e., the product of the crustal abundance of 244Pu and the enrichment factor of Ce (3.03 × 103, measured by XRF analysis) (Table 3). Thus, the upper detection limit (UDL) of 244Pu in 450 g rare earth concentrate (2.1 × 10−20 g/g) is still ~18 folds higher than the expected 244Pu content in this bastnaesite according to the initial (244Pu/238U)ESS abundance ratio of 0.008. The UDL still needs to be pushed down further to detect the primordial 244Pu and to verify whether or not the initial abundance ratio is correct. However, it would be a great analytical challenge as 8 kg of the present bastnaesite concentrate sample is required to be processed. With such an increase of sample volume, it is difficult to ensure the high chemical recovery and low background of Pu.
|
|
Table 3 Estimated upper limits of 244Pu refer to the enrichment factor of Th and Ce in the bastnaesite mineral. |
{kind=link}
{kind=link}
According to genetic mineralogy and minerocoenology, mineral symbiosis follows the principle of isomorphism, which means that the positions of some ions, atoms or molecules in the crystal structure are occupied by the others with similar properties. Compared to Ce4+ and Ce2O3, the radius of Pu4+ ion is even closer to that of the Th4+ ion, and the melting point of PuO2 is also similar to that of ThO2. Therefore, the behavior of Pu in rare earth minerals may be more similar to Th in the long process of geological evolution. Then, the expected 244Pu content in this bastnaesite sample is calculated to be 6.5 × 10−23 g/g by multiplying the upper crustal abundance of 244Pu with the enrichment factor of Th (162, measured by XRF analysis [11]). As the upper detection limit of 244Pu achieved for 450 g rare earth concentrate is 2.1 × 10−20 g/g, the direct detection of primordial 244Pu in this sample seems insurmountably difficult. The enrichment factor of plutonium in any rare earth mineral seems unlikely to significantly exceed the maximum enrichment of Th (2 × 107) and rare earth elements (3 × 105) in its richest mineral [9]. Other samples with high concentrations of Th, such as monazite and gadolinite concentrate, would probably be the best candidates for a positive detection of primordial 244Pu in nature. Alternatively, with more detailed studies of the differentiation ratio of Pu/Th in rare earth extraction process for correction of the content of primordial 244Pu, sample volume could be reduced by analyzing the highly concentrated Th residue samples from processing monazite for extraction of REEs.
In summary, the measurement of 244Pu in the HCl leachate and residue of 450 g of Bayan Obo bastnaesite concentrate was performed using the complete digestion ultra-sensitive analytical method, for attempting to verify the initial (244Pu/238U)ESS abundance ratio in the early formation of the solar system. Attogram level of 239Pu was found in the sample target, and the counting rate of 239Pu in the residue was ~4.5 times higher than that in the HCl leachate, which imply that Pu could preferentially concentrate in its tetravalent state with Th(IV) in the bastnaesite. Since no true signal of 244Pu was observed, an upper detection limit for the 244Pu content in our bastnaesite concentrate sample was estimated to be 2.1 × 10−20 g/g at 99% confidence level, which is the lowest upper limit obtained so far. This result denies the discovery of 1.0 × 10−18 g/g primordial 244Pu in bastnaesite reported by Hoffman and Lawrence. The possible upper limit of 244Pu content in our sample was estimated to be in the range of 1.2 × 10−21 g/g and 6.5 × 10−23 g/g by assuming the same enrichment factor of Pu as cerium or thorium, respectively. Therefore, it would still be problematic for direct search of primordial 244Pu in terrestrial samples even if a much larger size of this sample is processed. Samples with high concentrations of Th could be the most promising choice for the future study.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentThis work was financially supported by the National Natural Science Foundation of China (Nos. 11675150, 12005197 and 12175201).
| [1] |
D.C. Hoffman, F.O. Lawrence, Nature 234 (1971) 132-134. DOI:10.1038/234132a0 |
| [2] |
S. Goriely, G. Martinez-Pinedo, Nucl. Phys. 944 (2015) 158-176. DOI:10.1016/j.nuclphysa.2015.07.020 |
| [3] |
J. Kunz, T. Staudacher, C.J. Allegre, Science 280 (1998) 877-880. DOI:10.1126/science.280.5365.877 |
| [4] |
G. Turner, A. Busfield, S. Crowther, et al., Earth Planet. Sci. Lett. 261 (2007) 491-499. DOI:10.1016/j.epsl.2007.07.014 |
| [5] |
A. Wallner, T. Faestermann, J. Feige, et al., Nat. Commun. 6 (2015) 1-9. DOI:10.1038/ncomms6956 |
| [6] |
P. Steier, E. Hrnecek, A. Priller, et al., Nucl. Nucl. Inst. Methods Phys. Res. B 294 (2013) 160-164. DOI:10.1016/j.nimb.2012.06.017 |
| [7] |
A. Wallner, M.B. Froehlich, M.A.C. Hotchkis, et al., Science 372 (2021) 742-745. DOI:10.1126/science.aax3972 |
| [8] |
G. Raisbeck, T. Tran, D. Lunney, et al., Nucl. Inst. Methods Phys. Res. B 259 (2007) 673-676. DOI:10.1016/j.nimb.2007.01.205 |
| [9] |
P.R. Fields, A.M. Friedman, J. Milsted, et al., Nature 212 (1966) 131-134. DOI:10.1038/212131a0 |
| [10] |
J. Lachner, I. Dillmann, T. Faestermann, et al., Phys. Rev. C 85 (2012) 1-6. DOI:10.1103/PhysRevC.85.015801 |
| [11] |
Y. Wu, X.X. Dai, M. Christl, et al., ACS Earth Space Chem. 5 (2021) 1316-1324. DOI:10.1021/acsearthspacechem.0c00288 |
| [12] |
E.C. Alexander, Science 172 (1971) 837-840. DOI:10.1126/science.172.3985.837 |
| [13] |
M. Li, X.W. Zhang, Z.G. Liu, et al., Rare Met. 32 (2013) 312-317. DOI:10.1007/s12598-013-0034-0 |
| [14] |
M. Christl, X.X. Dai, J. Lachner, et al., Nucl. Inst. Methods Phys. Res. B 331 (2014) 225-232. DOI:10.1016/j.nimb.2013.11.045 |
| [15] |
M. Christl, C. Vockenhuber, P.W. Kubik, et al., Nucl. Inst. Methods Phys. Res. B 294 (2013) 29-38. DOI:10.1016/j.nimb.2012.03.004 |
| [16] |
C. Vockenhuber, V. Alfimov, M. Christl, et al., Nucl. Inst. Methods Phys. Res. B 294 (2013) 382-386. DOI:10.1016/j.nimb.2012.01.014 |
| [17] |
X.X. Dai, M. Christl, S. Kramer-Tremblay, et al., Anal. Chem. 88 (2016) 2832-2837. DOI:10.1021/acs.analchem.5b04546 |
| [18] |
H.A. Synal, Int. J. Mass Spectrom. 349-350 (2013) 192-202. DOI:10.1016/j.ijms.2013.05.008 |
| [19] |
K.M. Wilcken, T.T. Barrows, L.K. Fifield, et al., Nucl. Inst. Methods Phys. Res. 259 (2017) 727-732. |
| [20] |
G.T. Seaborg, M.L. Pearlman, J. Am. Chem. Soc. 70 (1948) 1571. DOI:10.1021/ja01184a083 |
| [21] |
C.A. Levine, G.T. Seaborg, J. Am. Chem. Soc. 73 (1951) 3276. DOI:10.1021/ja01151a085 |
| [22] |
C. Tighe, M. Castrillejo, M. Christl, et al., Nat. Commun. 12 (2021) 1381. DOI:10.1038/s41467-021-21575-9 |
| [23] |
G.J. Feldman, R.D. Cousins, Phys. Rev. D 57 (1998) 3873-3889. DOI:10.1103/PhysRevD.57.3873 |
| [24] |
K.H. Wedepohl, Cosmochim. Acta 59 (1995) 1217-1232. DOI:10.1016/0016-7037(95)00038-2 |
| [25] |
K. Holliday, T. Hartmann, K.J. Czerwinski, Nucl. Mater. 392 (2009) 487-493. DOI:10.1016/j.jnucmat.2009.04.013 |
| [26] |
J.H. Jones, D.S. Burnett, Geochim. Cosmochim. Acta 51 (1987) 769-782. DOI:10.1016/0016-7037(87)90091-3 |