 2022, Vol. 33
2022, Vol. 33 

b Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China;
c Shanghai Industrial Technology Research Institute (SITRI), Shanghai 201800, China;
d School of Stomatology, Dalian Medical University, Dalian 116044, China;
e State Key Laboratory of component-based Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin 301617, China;
f Institute of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin 301617, China
Drug screening usually costs more than 10 years, and over 90% drugs that succeed in the animal experiments showed liver inject during clinic test [1-3]. Therefore, an excellent drug screening model showed as similar reaction of the drugs as human beings could not only promote drug discovery period, but also have a great influence on the development of drug screening. Till now in vivo animal models were the essential way for the drug screening, however, species difference was its most prominent shortcoming and the main reason to cause the high failure rate of drug screening [4-10]. Moreover, the in vivo models are difficult to study the biological processes and signal paths in the in vivo animal models.
In this case, it is an urgent requirement to develop an in vitro liver model which would show the same result as the clinic test so that it would be able to be used for the study of the processes of how the drugs induce the injury of liver and caused the liver diseases. The liver is the main site of drug metabolism, and the key functional unit of liver is lobule [11-16]. In vivo, the drug enters the liver through sinusoid, a special capillary, which consists of the endotheliocytes. Its main function is carrying out material exchange between liver cells and blood stream. Drugs filtrated from the blood can pass through the sinusoid into the liver and then metabolize under the action of P450 enzymes. The drugs may produce some toxicity induce the injury of liver, generally called drug-induced liver injury (DILI).
Currently, several kinds of the in vitro liver models were developed, [17-22] such as traditional 2D cultured hepatocytes, 3D-printed liver tissue [23], liver organoids [24], and liver-on-a-chip systems [25]. Bhise et al. [26] developed a long-term cultured liver chip with HepG2 cells or C3A cells. They used the methacryloyl hydrogel to wrap around the HepG2 cells or C3A cells to form spheroids, and then, the spheroids were arranged in the microfluidic chip by the bio-printer. The spheroids could be alive for 30 days which would be applied in chronic toxicity test. Massa et al. [27] designed a 3D liver with vascular layer by integrating hydrogel scaffold, 3D bio-printing and microfluidic technology, and the experiment proved that the platform can effectively simulate the hepatic vascular layer, which is conducive to observe and predict the mechanism of drug hepatotoxicity at the microcirculation level. Kostadinova et al. [28] established a nylon scaffold that allowed three-dimensional culture of liver cells and non-parenchymal cells, including kuffer cells, stellate cells, hepatic sinusoidal endothelial cells and bile duct endothelial cells, which could maintain the specific function of liver for a long time and form a cholangiole structure in response to inflammatory stimuli. Zuchowska et al. [29] tested hepatotoxicity of 5-fluorouracil (5-FU), an anticancer drug, in HepG2 cells based on long-term 3D spheroid culture in microfluidic system, and found that resistance of HepG2 cells to 5-FU of two tested concentrations decreased with the increase of spheroid diameter. For each model, there are respective features, however, it is well known that liver-on-a-chip technology is innovative to manage liver microenvironments in vitro [30-35], and 3D-printed liver tissue could offer the cells a 3D culture condition which is as same as in vivo. Therefore, the in vitro model combining the microfluidic system with 3D bio-printing technology could simulate the in vivo tissue function better.
Here we present a transwell inserted polymethyl methacrylate (PMMA) chip which could fit the 3D bioprinter to construct a perfusion liver-on-a-chip by co-cultured the human liver tumor cell line (HepG2) and human umbilical vein endotheliocyte (HUVEC). HUVECs were cultured on the bottom of the tanswells’ membrane with perfusion to mimic the share force in vivo, and HepG2 cells were seeded on the top of the transwells’ membrane by the bioprinter and cultured in 3D condition to keep the cell growth situation closer to the in vivo. Therefore, the cells could maintain the better liver cell functions such as albumin secretion, urea synthesis and drug metabolism enzyme P450 expression for at least 14 days, which means this platform could be used in chronic toxicity test. We designed the size of the chip as same as the 96 well plates, so that it could be compatible to various detecting instruments such as microplate reader and microscope carrier. Moreover, the open design of the chip can not only to adapt the bioprinter but also facilitate the subsequent testing. Compare to the traditional polydimethyl siloxane (PDMS) chip, PMMA is more suitable for large-scale production to avoid the lot-to-lot variation. Additionally, this chip is reusable by replacing the transwells only, so that the cost could be decreased significantly and it is possible to commercialize.
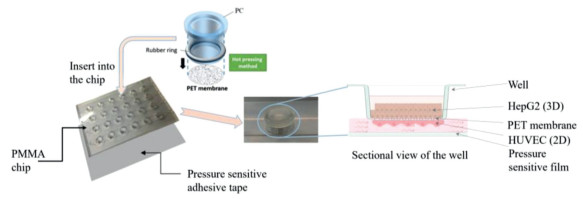
The structure of the chip was shown in Fig. 1, it consisted of three parts include wells, chip with compartment and channels, pressure sensitive adhesive tape. 24 compartments arranged in rows above 4 channels, the pressure sensitive adhesive tape was adhered on the bottom of the chip to seal the channels, and then wells were inserted into each compartment and apart into two parts, as shown in Fig. 1. Before cells seed, the chip would be sterilized by the autoclave. Place the chip upside down, and then 20 μL HUVEC suspension with density of 5 × 105 cells/mL was added on the bottom of the membrane in each well cultured for 24 h after the adherence, then the chip was turned over. After that 1 × 106 cells/mL HepG2 cell suspension was printed on the top of the membrane which precoated with the mixture of collagen and matrigel at the ratio of 1:1 in order to keep the printed cells flock together. The setting of the printer is 100 times spraying, 10 times repeat, 4 spray heads worked, and the print height is 5 mm. The printed droplet contains 600â€"800 cells, and its diameter is about 400 μm, as shown in Figs. 2a and b. At last, HUVEC perfused by the peristaltic pump with the speed of 99 μL/min as shown in Figs. 2c and d. The two kinds of cells were co-cultured in the chip for at least 14 days, and the cell viability was tested with calcein-AM (live cells) and ethidium homodimer-1 (dead cells) (live/dead viability/cytotoxicity kit, Invitrogen, L3224) and also, the ATP of the cells was tested by the CellTiter-GloⓇ Luminescent Cell Viability Assays (Promega, G7570). The tight conjunction of HUVEC represented by the immunofluorescent staining of tight junction protein VE-cadherin and ZO-1, and the quantitative results of cells cultured in static condition and perfusion condition were compared. The cell functions of HepG2 included albumin secretion, urea synthesis, drug metabolism enzyme P450 expressed by the immunofluorescent staining and the test kit (BCP Albumin Assay Kit, sigma), and the results of HepG2 spheroids were compared to the 2D cultured HepG2 cells. After the two types of cells seeded in the chip and cultured for several days we verified the drug toxicity function of the liver chip by acetaminophen (APAP). After 14 days cultured, the HepG2 spheroids were exposed in 0.0, 1.0, 2.0, 3.0, 4.0, 5.0 mmol/L of APAP solution for 24 h, and then the reactive oxygen species (ROS) were tested by the MitoTrackerâ„¢ Red CMXRos (Thermo Fisher), the albumin secretion and urea synthesis were assayed by the test kit mentioned before. All the fluorescent quantitation was assayed by the Image J.

|
Download:
|
| Fig. 1. The chip structure diagram of the chip. | |
{kind=link}

|
Download:
|
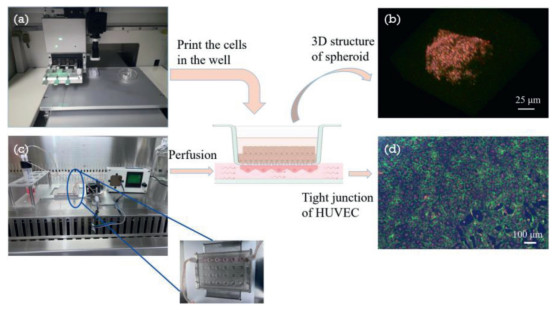
| Fig. 2. The schematic diagram of cell culture in the chip. (a) Cells seeded in the topside of the chip by the bio-printer, (b) photo of spheroid 3D structure taken by the confocal microscope, (c) perfusion of the HUVEC by peristaltic pump, (d) immunofluorescence image of VE-cadherin (green), F-actin(red) and DAPI (blue) which shown the tight junction of HUVEC after perfusion. | |
{kind=link}
In order to verify the function of the liver-on-chip model, first of all we expressed the viability of the two cells after 14 days culture. As shown in Fig. S1a (Supporting information), above 95% area of the HUVECs maintained alive (green), and at least 85% area of the HepG2 cells were alive. Since the cells inside spheroids were difficult to be observed by the live/dead stain method, we detected the ATP of them. As the result which showed in Fig. S1b (Supporting information), the ATP levels of 14 days cultured spheroids were higher than that of 3 days cultured spheroids because the cell number was increased due to the cell proliferation and it also means the cell viability maintained good. Perfusion offered HUVEC the share force as same as in vivo, and moreover it would bring the fresh media to the cells, at the same time it also took away the wastes and the dead cells. This was thought to be the main reason why HUVECs could keep good viability after 14 days cultured. The HepG2 cells suspension droplets were printed on the membrane of the transwells and then formed spheroid and the spheroid morphological structure of the HepG2 was observed by the confocal microscope as shown in Fig. 2c which proved that the cells were grown in a 3D condition as same as in vivo. The 3D cultured condition was thought to be the reason why the viability of the spheroids was well maintained for a long term. It also indicated that the long-term co-culture of the two types of cells, HepG2 and HUVEC, had realized in the chip.
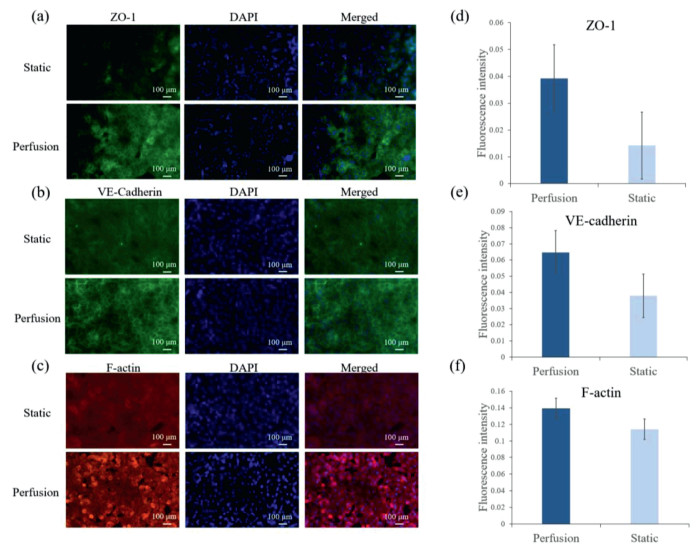
The most important factor for a vitro model is that if it could maintain the cell functions as well as in vivo or not. Microenvironment, which includes extracellular matrix (ECM), intercellular interaction, physical force and so on, plays an important role in cell growth and function maintenance. We assayed the expression of ZO-1, VE-cadherin and F-actin expression with 7 days perfusion and them in static condition. The results showed that the expression of all three proteins was higher than them in the static condition as shown in Fig. 3. That was because the perfusion offered the shear force which could make HUVEC connect tightly and form stress fiber, so the HUVEC could perform the barrier function to allow substances through selectively.

|
Download:
|
| Fig. 3. Tight junction of the HUVEC were characterized by immunofluorescence of tight junction protein: (a) ZO-1, (b) VE-cadherin, (c) F-actin, and (d) corresponding fluorescent quantitation results of ZO-1, (e) corresponding fluorescent quantitation results of VE-cadherin, (f) corresponding fluorescent quantitation results of F-actin. | |
{kind=link}
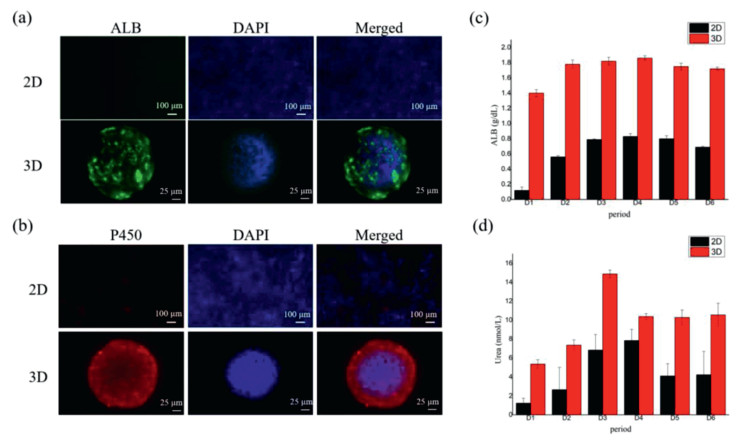
The verification of liver physiological function could be characterized by albumin (ALB) secretion, urea synthesis, drug metabolism enzyme P450 expression. Albumin is secreted by liver, and its function is to maintain the plasma colloid osmotic pressure constant. Urea synthesis signified the metabolism of amino acid which is another important function of liver. P450 enzyme expression means the drug metabolism function of liver. Therefore, we indicated these characterized markers of liver expression of HepG2 spheroids and the results were compared with the cells culture in 2D condition as shown in Figs. 4a and b. Moreover, albumin and urea were quantified by the assay kits. As shown in Figs. 4c and d, both the secretion of the ALB and the synthesis of urea of the HepG2 cells in chip were much higher than that of the cells in 2D cultured condition. That means the satiation and function of the cells cultured in 3D condition were more similar to the in vivo condition.

|
Download:
|
| Fig. 4. Liver physiological function characterization of the HepG2 cells cultured in the chip. (a) Immunofluorescence photo of ALB, (b) immunofluorescence photo of P450 (Red), (c) corresponding fluorescent quantitative analysis of ALB, (d) corresponding fluorescent quantitative analysis of Urea. | |
{kind=link}
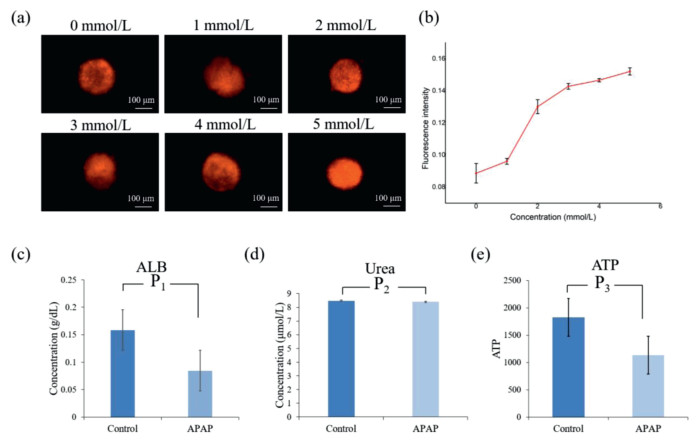
After confirming the successful construction of the liver physiological model, we applied it to the study of liver injury with drugs. Acute liver failure (ALF) caused by these factors can lead to extensive necrosis or apoptosis of liver cells, inflammatory reaction of hepatocyte steatosis, oxidative stress, and even liver function impairment. It is a serious disease with high mortality. Acetaminophen (APAP) is a commonly used antipyretic, drug abuse or accidental overdose is the most common cause of acute liver injury. APAP plays a dominant role in the acute liver injury caused by drug poisoning. Researches showed that excessive APAP in the liver through a series of reactions, will produce a large number of reactive oxygen species (ROS) cause serious oxidative damage of the liver, and eventually lead to acute liver injury. Therefore, we treated the HepG2 spheroids with different concentrations of APAP for 24 h, after that, the ROS would be quantity assayed by MitoTrackerâ„¢ Red CMXRos, and the ALB secrete, Urea synthesis was also detected. Moreover, after the treatment of APAP we detected the ATP, in order to get the cell viability.
We investigated the production of ROS in HepG2 cells after being exposed to APAP and the results were shown in Figs. 5a and b, APAP significantly induced the generation of ROS in HepG2 cells and this effect exhibited a dose-dependent manner. In addition, according to the quantitative results using microplate reader assay, 4 mmol/L of APAP would significantly reduce the albumin expression, while would not impact the synthesis of urea, as shown in Figs. 5c and d. And the cell viability reduced after the treatment of 4 mmol/L APAP as shown in Fig. 5e. This was thought to be the reason why the expression of albumin was descended.

|
Download:
|
| Fig. 5. (a) ROS production in HepG2 cells after exposure to different concentrations of APAP, (b) corresponding fluorescent quantitation results of ROS. (c) Quantitative analysis result of ALB, (d) Urea and (e) ATP before and after the treatment of 4 mmol/L APAP using a microplate reader. | |
{kind=link}
In conclusion, we proposed a demountable liver on chip device for study of liver drug toxicity. The proportion and the spatial structure of HepG2, HUVEC cells were integrated in an orderly manner, which simulated the complex structure and microenvironment of liver in vivo, and the process of liver injury caused by APAP also reappeared through this device. The results showed that this liver-device is able to recreate the damage process of hepatic cells induced by APAP, while measuring multiple biomarkers of hepatic cells to understand the intercellular communication between each other. Thus, this in vitro liver-on-chip device could serve as a functional platform for drug-induced hepatotoxicity, liver function and disease. Moreover, this chip could also be used to rebuild other organ in vitro such as brain blood barrier which is studied by our team and so on. By culture different kinds of cells in the chip, several organs on a chip would develop in this PMMA chip, and it could be widely used in the drug toxicity test field.
Declaration of competing interestThe authors declare that they do not have any known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors are grateful for the support of grants from The National Key R & D Program of China (No. 2018YFA0108202), National Science Foundation, China (Nos. 61971410, 61801464, 62001458 and 61801465), Shanghai Sailing Program (No. 20YF1457100), China Postdoctoal Science Foundation (No. 2020000246), Shanghai Engineer & Technology Research Center of Internet of Things for Respiratory Medicine (No. 20DZ2254400) and the Science and Technology Commission of Shanghai Municipality (No. 19511104200).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.029.
| [1] |
J.A. Dimasi, R.W. Hansen, H.G. Grabowski, J. Health Econ. 22 (2003) 151-185. DOI:10.1016/S0167-6296(02)00126-1 |
| [2] |
J.D. Caplin, N.G. Granados, M.R. James, Adv. Healthcare Mater. 10 (2015) 1426-1450. DOI:10.1002/adhm.201500040 |
| [3] |
R.J. Church, P.B. Watkins, Liver Int. 37 (2017) 1582-1590. DOI:10.1111/liv.13441 |
| [4] |
R.J. Church, G.A.K. Ublick, J. Aubrecht, et al., Hepatology 69 (2019) 760-773. DOI:10.1002/hep.29802 |
| [5] |
M.R. Mcgill, M.R. Sharpe, C.D. Williams, et al., Clin. Invest. 4 (2012) 1574-1583. DOI:10.1172/JCI59755 |
| [6] |
J.W. Haycock, 3D cell culture: a review of current approaches and techniques, in: J. Haycock (Ed. ), 3D Cell Culture. Methods in Molecular Biology (Methods and Protocols), Humana Press, 2011, pp. 1-15.
|
| [7] |
M.A. Lancaster, J.A. Knoblich, Science 345 (2014) 1247125. DOI:10.1126/science.1247125 |
| [8] |
S. Knowlton, S. Tasoglu, Trends Biotechnol. 9 (2016) 681-682. DOI:10.1016/j.tibtech.2016.06.005 |
| [9] |
R.R. Zhang, M. Koido, T. Tadokoro, et al., Stem Cell Rep. 10 (2018) 780-793. DOI:10.1016/j.stemcr.2018.01.006 |
| [10] |
M. Jang, P. Neuzil, T. Volk, et al., Biomicrofluidics 9 (2015) 034113. DOI:10.1063/1.4922863 |
| [11] |
E. Tinois, J. Tiollier, M. Gaucherand, et al., Exp. Cell. Res. 193 (1991) 310-319. DOI:10.1016/0014-4827(91)90102-Z |
| [12] |
J.N.W.N. Barker, C.E.M. Griffiths, B.J. Nickoloff, et al., Lancet 337 (1991) 211-214. DOI:10.1016/0140-6736(91)92168-2 |
| [13] |
R.C. McKenzie, D.N. Sauder, J. Invest. Dermatol. 95 (1990) S105-S107. DOI:10.1111/1523-1747.ep12874955 |
| [14] |
J. Varani, P. Perone, D.M. Spahlinger, et al., Toxicol. Pathol. 35 (2007) 693-701. DOI:10.1080/01926230701481907 |
| [15] |
O.N. Bae, S. Ahn, S.H. Jin, et al., Toxicol. Appl. Pharmacol. 283 (2015) 147-155. DOI:10.1016/j.taap.2015.01.008 |
| [16] |
M. Lee, J.H. Hwang, K.M. Lim, Toxicol. Res. 33 (2017) 191-203. DOI:10.5487/TR.2017.33.3.191 |
| [17] |
A.A. Banaeiyan, J. Theobald, J. Paukštyte, et al., Biofabrication 9 (2017) 015014. DOI:10.1088/1758-5090/9/1/015014 |
| [18] |
J. Zhu, Z.Q. Zhang, Y.T. Zhang, et al., Biochem. Biophys. Res. Commun. 496 (2018) 176-183. DOI:10.1016/j.bbrc.2018.01.019 |
| [19] |
M.H. Forouzanfar, A. Afshin, L.T. Alexander, et al., The Lancet 388 (2016) 1659-1724. DOI:10.1016/S0140-6736(16)31679-8 |
| [20] |
Y.A. Lee, M.C. Wallace, S.L. Friedman, Gut 64 (2015) 830-841. DOI:10.1136/gutjnl-2014-306842 |
| [21] |
D. Bovard, A. Iskandar, K. Luettich, et al., Toxicol. Res. Appl. 1 (2017) 1-16. DOI:10.1177/2397847317726351 |
| [22] |
K.M. Jung, S.H. Lee, W.H. Jang, et al., Toxicol. In Vitro 28 (2014) 742-750. DOI:10.1016/j.tiv.2014.02.014 |
| [23] |
J. Varani, P. Perone, D.M. Spahlinger, et al., Toxicol. Pathol. 35 (2007) 693-701. DOI:10.1080/01926230701481907 |
| [24] |
A. Coquette, N. Berna, A. Vandenbosch, et al., Toxicol. In Vitro 17 (2003) 311-321. DOI:10.1016/S0887-2333(03)00019-5 |
| [25] |
H.Y. Lee, M. Stieger, N. Yawalkar, et al., Mediators Inflammation 2013 (2013) 916497. DOI:10.1155/2013/916497 |
| [26] |
N.S. Bhise, V. Manoharan, S. Massa, et al., Biofabrication 8 (2016) 014101. DOI:10.1088/1758-5090/8/1/014101 |
| [27] |
S. Massa, M.A. Sakr, J. Seo, et al., Biomicrofluidics 11 (2017) 044109. DOI:10.1063/1.4994708 |
| [28] |
R. Kostadinova, F. Boess, D. Applegate, et al., Toxicol. Appl. Pharmacol. 268 (2013) 1-16. DOI:10.1016/j.taap.2013.01.012 |
| [29] |
A. Zuchowska, K. Kwapiszewska, M. Chudy, et al., Electrophoresis 38 (2017) 1206-1216. DOI:10.1002/elps.201600417 |
| [30] |
I.R. Williams, T.S. Kupper, Life Sci. 58 (1996) 1485-1507. DOI:10.1016/0024-3205(96)00042-2 |
| [31] |
O.N. Bae, M. Noh, Y.J. Chun, et al., Biomol. Ther. 23 (2015) 12-18. DOI:10.4062/biomolther.2014.102 |
| [32] |
D.M. Bagley, D. Waters, B.M. Kong, Food Chem. Toxicol. 32 (1994) 1155-1160. DOI:10.1016/0278-6915(94)90131-7 |
| [33] |
N.P. Luepke, Food Chem. Toxicol. 23 (1985) 287-291. DOI:10.1016/0278-6915(85)90030-4 |
| [34] |
L. Gilleron, S. Coecke, M. Sysmans, et al., Toxicol. In Vitro 10 (1996) 431-446. DOI:10.1016/0887-2333(96)00021-5 |
| [35] |
J.H. Fentem, D. Briggs, C. Chesne, et al., Toxicol. In Vitro 15 (2001) 57-93. DOI:10.1016/S0887-2333(01)00002-9 |