 2022, Vol. 33
2022, Vol. 33 


b Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou 510530, China;
c Key Laboratory of Jiangxi Province for Persistent Pollutants Control and Resources Recycle, Nanchang Hangkong University, Nanchang 330063, China
Pyridine is one of the most prevalent heterocyclic skeletons in bioactive molecules. The fluorine atom embodies high electronegativity and a small atomic radius [1, 2], and its incorporation into pyridine drastically improves the lipophilicity, bioavailability, metabolic stability and, in some cases, the potency of known biologically active molecules (Fig. 1). Despite their increasing importance in pharmaceuticals and agrochemicals [3-8], developing a simple and practical method to prepare fluoropyridines, especially 4-fluoropyridines, still encounters great challenges.

|
Download:
|
| Fig. 1. Selected biologically active 4-fluoropyridines. | |
The classical approach for the formation of 4-fluoropyridines is the Balz-Schiemann reaction (Scheme 1a) [9]. Acidic conditions, toxicity of the reagents, and potential for explosions impede their synthetic applications. Nucleophilic substitution [10-20] of 4-halo or nitropyridines by inorganic or organic fluoride salts [21-31] offers an alternative route for accessing 4-fluoropyridines (Scheme 1b). However, the poor solubility of inorganic fluoride salts, stoichiometric usage of cost and toxic organic fluoride salts, make the reaction proceed under harsh conditions, leading to inferior conversion and functional group tolerance.

|
Download:
|
| Scheme 1. Tactics for the construction of 4-fluoropyridines. | |
gem-Difluorocyclopropene is an intriguing framework, as it contains three important structural motifs: electrophilic difluoromethylene, alkene and the smallest carbocycle, thus possessing unusual electric and steric properties. Taking advantage of the gem-difluorocyclopropene as a valuable two-carbon donor, Waser and coworkers reported a [3 + 2] annulation of cyclopropenes with cyclopropylanilines to access to 6, 6-difluorobicycle[3.1.0]hexanes [32]. Furthermore, Cossy realized a concise, and efficient synthesis of 5-fluoropyridazines by a [3 + 3] cycloaddition reaction [33] using the gem-difluorocyclopropene as a three-carbon donor [34, 35]. Considering the importance of 4-fluoropyridines in pharmaceuticals and agrochemicals, we herein report that a convenient and effective [3 + 2]/[2 + 1] annulation reaction between gem-difluorocyclopropenes [36-49] and isocyanides 2 [50-55] or aldimine esters 3 [56-59] to produce highly functionalized 4-fluoropyridines in moderate to good yields (Scheme 1c). Specifically, the whole transformation is characterized by the following points: i) Unlike the direct fluorination reactions of pyridines, this novel cycloaddition reaction originates from readily available substrates, and the construction of the pyridine ring and the introduction of a fluorine atom at specific position are completed simultaneously, thus providing a new pathway for the synthesis of 4-fluoropyridines; ii) In the cyclization reactions, the carbon-carbon π bond of gem-difluorocyclopropene is broken, while two carbon-carbon single bonds are formed; iii) The reactions are conducted under mild and air conditions, by which both tri- and tetra-substituted 4-fluoropyridines are obtained in moderate to good yields with wide substrate applicability and pure regioselectivity; iv) In the synthesis of tetra-substituted 4-fluoropyridines, the reaction proceeds without transition metals, and DMSO is utilized as both a solvent and an oxidant.
The reaction was initially investigated using gem-difluorocyclopropene 1a and ethyl isocyanoacetate 2a as model substrates, AgOAc as the catalyst and K2CO3 as the base in PhCF3 at room temperature; to our delight, the desired product 4a was obtained in 91% yield (Table 1, entry 1). Other catalysts such as Ag2CO3, Ag2O, AgBF4 and Cu(OAc)2 prevented the substrate 1a from being completely converted, and the reaction did not proceed without the catalyst (Table 1, entries 2–6). Replacing K2CO3 with other bases made the reaction worse (Table 1, entries 7–9). Subsequent solvent screening revealed that PhCF3 made better conversion (Table 1, entries 10–12). Then the reaction between gem-difluorocyclopropene 1a and aldimine ester 3a in the conditions of AgOAc as the catalyst, K2CO3 as the base, DDQ as an oxidant, and PhCF3 as the solvent was detected. When the temperature was elevated to 70 ℃, tetra-substituted 4-fluoropyridine 5a was produced in 22% yield (Table 1, entry 13). Substituting K2CO3 with K3PO4 increased the yield to 41% (Table 1, entry 14). The reaction still conducted smoothly without the catalyst and gave the product 5a in 54% yield (Table 1, entry 15). When the solvent was changed to DMSO, the yield of 5a was further promoted to 65% (Table 1, entry 16). Interestingly, the reaction was almost impervious even if no DDQ was added (Table 1, entries 17 and 18). Consequently, the optimal reaction conditions for the preparation of 4a were determined to be 1a (0.2 mmol) in the presence of 2a (2.0 equiv.), AgOAc (10 mol%), K2CO3 (2.5 equiv.) and, PhCF3 (1 mL) at room temperature for 3 h, while 5a was obtained in the presence of 1a (0.2 mmol), 3a (1.5 equiv.), K3PO4 (1.5 equiv.) and DMSO (1 mL) at 70 ℃ for 4 h.
|
|
Table 1 Optimization of reaction conditionsa |

{kind=link}
{kind=link}
With optimized conditions in hand, the reactions between gem-difluorocyclopropenes 1 and isocyanides 2 were tested first. As shown in Scheme 2, the gem-difluorocyclopropene substrates bearing electron-donating substituents such as Me, Ph and electron-withdrawing substituents such as F, Cl, Br, CF3, NO2, CO2Me in the para- or meta-positions of the phenyl moiety worked smoothly under standard conditions, and gave the products 4a-4k in good yields. However, when the para position of the phenyl moiety in the gem-difluorocyclopropene substrate embedded strong electron-donating methoxy group, the yield of the product 4h was significantly reduced. Specifically, a 2-methyl-substituted substrate did not retard the process and gave product 4m in 87% yield. The substrates with aromatic heterocycles, for example, pyridine, quinolone and benzoxazole were also tolerant, producing the products 4p-4r in 68%−96% yields. Alkyl-substituted difluorocyclopropenes were also compatible with the reaction system. Methylene groups with nitrogen-centred linkages such as carbazole gave the product 4s in 81% yield, while oxygen-centred linkages such as naphthol afforded product 4t in 74% yield. The reaction still proceeded effectively with other esters such as methyl ester, tert-butyl ester, and benzyl ester, even amides in isonitriles (4u-4y), although the yields were slightly reduced. To further highlight the synthetic versatility of our method, several complex natural products and drugs, were subjected to the reaction conditions. Estetrol (1z) andl-(-)-borneol (1ab) derivatives gave the corresponding 4-fluoropyridines 4z and 4ab in 66% and 51% yield, respectively. Febuxostat 1aa and Adapalene 1ac also were converted to 4-fluoropyridine derivatives (4aa, 57% and 4ac, 45%).

|
Download:
|
| Scheme 2. Substrate scope for trisubstituted 4-fluoropyridines. Reaction conditions: 1 (0.2 mmol), 2 (2.0 equiv.), AgOAc (10 mol%), K2CO3 (2.5 equiv.), PhCF3 (1 mL) at room temperature for 3 h in air, isolated yield. | |
{kind=link}
The substrate scope for tetra-substituted 4-fluoropyridines was then screened (Scheme 3). The gem-difluorocyclopropene substrates with both electron-donating substituents such as Me, Ph, OMe and electron-withdrawing substituents such as Br in the para- or meta-positions of the phenyl moiety were utilized under standard conditions, and generated the products 5a-5d in moderate to good yields. Subsequent screening of the aldimine esters [60] revealed that electrical properties of the substituted groups in the para- or meta-positions of the benzene ring had little effect on the reaction (5f-5j). Other esters such as ethyl ester were also compatible with the reaction, producing the product 5l in 50% yield. Out of our expectation, aldimine esters with alkyl groups such as benzyl, n-pentyl, n-butyl, n-propyl, and even alkenyl and ester groups showed no reactivity in the protocol.

|
Download:
|
| Scheme 3. Substrate scope for tetra-substituted 4-fluoropyridines. Reaction conditions: 1 (0.2 mmol), 2 (1.5 equiv.), K3PO4 (1.5 equiv.), DMSO (1 mL) at 70 ℃ for 4 h in air. Isolated yields. | |
{kind=link}
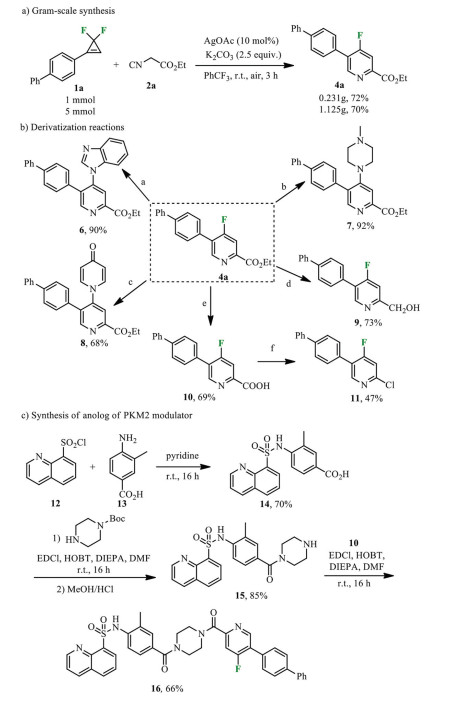
To demonstrate the scalability of this methodology, the reaction of 1a with 2a was performed on 1.0 mmol and 5.0 mmol scales, affording 72% and 70% yields of the product 4a, respectively (Scheme 4a). The nucleophilic substitutions of 4-fluoropyridine 4a with benzimidazole, 1-methylpiperazine and 4-hydroxypyridine released the corresponding products 6, 7 and 8 in 90%, 92% and 68% yield, respectively (Scheme 4b, paths a-c). NaBH4 reduction of the carbonyl group in 4a afforded alcohol 9 in 73% yield (path d). Hydrolysis of the ester groups in 4a produced 10 in 69% yield (path e), which further conducted a chlorinated decarboxylation reaction to obtain the product 11 in 47% yield (path f). Next, total syntheses of the anolog of PKM2 modulator was investigated (Scheme 4c). The product 15 was formed after a two-step reaction sequence of the sulfamidation and condensation, which further condensed with 10 to finally obtain the anolog of PKM2 modulator 16.

|
Download:
|
| Scheme 4. Synthetic applications. Reaction conditions: (a) 4a (0.2 mmol), benzimidazole (0.4 mmol), MeCN (1 mL), 80 ℃, 4 h, Ar; (b) 4a (0.2 mmol), 1-methylpiperazine (0.4 mmol), MeCN (1 mL), r.t., 4 h, Ar; (c) 4a (0.2 mmol), 4-hydroxypyridine (0.4 mmol), K2CO3 (0.4 mmol), NMP (1 mL), at 140 ℃ for 3 h; (d) 4a (0.2 mmol), NaBH4 (0.6 mmol), ethanol (1 mL), 0–25 ℃, 2 h; (e) 4a (0.2 mmol), KOH (0.4 mmol), methanol (1 mL), 35 ℃, 2 h; (f) 4a (0.2 mmol), NaHCO3 (0.2 mmol), t-BuCl (0.3 mmol), CH2Cl2 (1 mL), 60 ℃. Isolated yield. | |
{kind=link}
On the basis of related literature reports [61, 62] and our preliminary studies, a plausible mechanism is proposed in Scheme 5. A [3 + 2] cycloaddition reaction of gem-difluorocyclopropenes 1 with isocyanides 2 or imines 3 to produce intermediates I and II. The synergy of the electron-pushing effect of α-carbon of the ester group and the electron-pulling effect of gem-difluoride stimulates the ring-opening defluorination reaction of intermediates I and II, releasing intermediates III and IV. Finally, the intermediate IV was oxidized by DMSO to generate tetrasubstituted-4-fluoropyridines 5.

|
Download:
|
| Scheme 5. Possible catalytic cycle. | |
{kind=link}
In summary, we report a novel [3 + 2]/[2 + 1] cycloaddition reaction of gem-difluorocyclopropenes with isocyanides or imines, providing a modular, concise and efficient approach for accessing tri- and tetra-substituted 4-fluoropyridines in moderate to good yields with high regioselectivity. This reaction starts from simple and readily available substrates, proceeding under mild and air conditions, and its substrate scope is general. In the synthesis of tetra-substituted 4-fluoropyridines, the reaction proceeds without transition metals, and DMSO is utilized as both a solvent and an oxidant.
Declaration of competing interestThe authors declare no conflict of interest.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21861007, 21702034), Natural Science Foundation of Guangxi Province (No. 2021GXNSFAA075024), "BAGUI Scholar" Program of Guangxi Province of China, High-Level Innovation Team and Distinguished Scholar Program in Guangxi Colleges and Universities, the Jiangxi Province Science and Technology Project (No. 20212BAB213024).
Supplementary materialsSupplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.031.
| [1] |
I. Ojima, J.R. McCarthy, J.T. Welch, Biomedical frontiers of fluorine chemistry, ACS Sympos. Ser., 639, American Chemical Society: Washington DC, 1996.
|
| [2] |
J.T. Welch, S. Eswarakrishanan, Fluorine in Bioorganic Chemistry. New York: John Wiley and Sons, 2001.
|
| [3] |
P. Jeschke, ChemBioChem 5 (2004) 570-726. DOI:10.1002/cbic.200300833 |
| [4] |
B.E. Smart, J. Fluorine Chem. 109 (2001) 3-11. DOI:10.1016/S0022-1139(01)00375-X |
| [5] |
P. Jeschke, Pest Manage. Sci. 66 (2010) 10-27. DOI:10.1002/ps.1829 |
| [6] |
D. O'Hagan, J. Fluorine Chem. 131 (2010) 1071-1081. DOI:10.1016/j.jfluchem.2010.03.003 |
| [7] |
S. SRPurser, P.R. Moore, S. Swallow, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320-330. DOI:10.1039/B610213C |
| [8] |
W.K. Hagmann, J. Med. Chem. 51 (2008) 4359-4369. DOI:10.1021/jm800219f |
| [9] |
H. Suschitzky, Adv. Fluorine Chem. 1 (1965) 4-12. |
| [10] |
P.A. Champagne, J. Desroches, J.D. Hamel, M. Vandamme, J.F. Paquin, Chem. Rev. 115 (2015) 9073-9174. DOI:10.1021/cr500706a |
| [11] |
M.G. Campbell, T. Ritter, Chem. Rev. 115 (2015) 612-633. DOI:10.1021/cr500366b |
| [12] |
J.W. Lee, M.T. Oliveira, H.B. Jang, et al., Chem. Soc. Rev. 45 (2016) 4638-4650. DOI:10.1039/C6CS00286B |
| [13] |
Y.Y. See, M.T. Morales-Colo´n, D.C. Bland, M.S. Sanford, Acc. Chem. Res. 53 (2020) 2372-2383. DOI:10.1021/acs.accounts.0c00471 |
| [14] |
D.J. Adams, J.H. Clark, Chem. Soc. Rev. 28 (1999) 225-231. DOI:10.1039/a808707e |
| [15] |
M.A. Lacour, M. Zablocka, C. Duhayon, J.P. Majoral, M. Taillefer, Adv. Synth. Catal. 350 (2008) 2677-2682. DOI:10.1002/adsc.200800428 |
| [16] |
D.W.J. Kim, S.T. Lim, M.H. Sohn, J.A. Katzenellenbogen, D.Y. Chi, J. Org. Chem. 73 (2008) 957-962. DOI:10.1021/jo7021229 |
| [17] |
P.S. Fier, J.F. Hartwig, J. Am. Chem. Soc. 134 (2012) 10795-10798. DOI:10.1021/ja304410x |
| [18] |
N. Ichiishi, A.J. Canty, B.F. Yates, M.S. Sanford, Org. Lett. 15 (2013) 5134-5137. DOI:10.1021/ol4025716 |
| [19] |
Y. Ye, S.D. Schimler, P.S. Hanley, M.S. Sanford, J. Am. Chem. Soc. 135 (2013) 16292-16295. DOI:10.1021/ja408607r |
| [20] |
C.M. Hong, A.M. Whittaker, D.M. Schultz, J. Org. Chem. 86 (2021) 3999-4006. DOI:10.1021/acs.joc.0c02845 |
| [21] |
H.R. Sun, S.G. DiMagno, J. Am. Chem. Soc. 127 (2005) 2050-2051. DOI:10.1021/ja0440497 |
| [22] |
S.D. Kuduk, R.M. DiPardo, M.G. Bock, Org. Lett. 7 (2005) 577-579. DOI:10.1021/ol047688v |
| [23] |
H. Sun, S.G. DiMagno, Angew. Chem., Int. Ed. 45 (2006) 2720-2725. DOI:10.1002/anie.200504555 |
| [24] |
P. Tang, W. Wang, T. Ritter, J. Am. Chem. Soc. 133 (2011) 11482-11484. DOI:10.1021/ja2048072 |
| [25] |
L.J. Allen, J.M. Muhuhi, D.C. Bland, R. Merzel, M.S. Sanford, J. Org. Chem. 79 (2014) 5827-5833. DOI:10.1021/jo5003054 |
| [26] |
S.D. Schimler, S.J. Ryan, D.C. Bland, J.E. Anderson, M.S. Sanford, J. Org. Chem. 80 (2015) 12137-12145. DOI:10.1021/acs.joc.5b02075 |
| [27] |
S.J. Ryan, S.D. Schimler, D.C. Bland, M.S. Sanford, Org. Lett. 17 (2015) 1866-1869. DOI:10.1021/acs.orglett.5b00538 |
| [28] |
S.D. Schimler, M.A. Cismesia, P.S. Hanley, et al., J. Am. Chem. Soc. 139 (2017) 1452-1455. DOI:10.1021/jacs.6b12911 |
| [29] |
M.A. Cismesia, S.J. Ryan, D.C. Bland, M.S. Sanford, J. Org. Chem. 82 (2017) 5020-5026. DOI:10.1021/acs.joc.7b00481 |
| [30] |
S.D. Schimler, R.D.J. Froese, D.C. Bland, M.S. Sanford, J. Org. Chem. 83 (2018) 11178-11190. DOI:10.1021/acs.joc.8b01762 |
| [31] |
M.T. Morales-Colón, Y.Y. See, S.J. Lee, et al., Org. Lett. 23 (2021) 4493-4498. DOI:10.1021/acs.orglett.1c01490 |
| [32] |
B. Muriel, A. Gagnebina, J. Waser, Chem. Sci. 10 (2019) 10716-10722. DOI:10.1039/c9sc03790j |
| [33] |
M.Y. He, Y.C. Chen, Y. Luo, et al., Green Synth. Catal. 1 (2020) 180-182. DOI:10.1016/j.gresc.2020.10.004 |
| [34] |
G. Tran, G.D. Pardo, T. Tsuchiya, et al., Org. Lett. 17 (2015) 3414-3417. DOI:10.1021/acs.orglett.5b01370 |
| [35] |
A. Feraldi-Xypolia, G. Fredj, G. Tran, et al., Asian J. Org. Chem. 6 (2017) 927-935. DOI:10.1002/ajoc.201700216 |
| [36] |
F. Wang, T. Luo, J. Hu, et al., Angew. Chem. Int. Ed. 50 (2011) 7153-7157. DOI:10.1002/anie.201101691 |
| [37] |
L. Li, F. Wang, C. Ni, J. Hu, Angew. Chem. Int. Ed. 52 (2013) 12390-12394. DOI:10.1002/anie.201306703 |
| [38] |
X.Y. Deng, J.H. Lin, J. Zheng, J.C. Xiao, Chem. Commun. 51 (2015) 8805. DOI:10.1039/C5CC02736E |
| [39] |
W. Xu, Q.Y. Chen, J. Org. Chem. 67 (2002) 9421-9427. DOI:10.1021/jo020431v |
| [40] |
Z.L. Cheng, Q.Y. Chen, J. Fluorine Chem. 126 (2005) 39-43. DOI:10.1016/j.jfluchem.2004.10.003 |
| [41] |
Z.L. Cheng, Q.Y. Chen, Chin. J. Chem. 24 (2006) 1219-1224. DOI:10.1002/cjoc.200690227 |
| [42] |
Z.L. Cheng, Q.Y. Chen, J. Fluorine Chem. 127 (2006) 894-900. DOI:10.1016/j.jfluchem.2006.03.020 |
| [43] |
T. Nihei, T. Hoshino, T. Konno, Org. Biomol. Chem. 13 (2015) 3721-3731. DOI:10.1039/C5OB00046G |
| [44] |
X. Zhao, S. Xu, Y. Zhou, S. Cao, Org. Chem. Front. 6 (2019) 2539-2543. DOI:10.1039/c9qo00580c |
| [45] |
K. Yamani, H. Pierre, A. Archambeau, C. Meyer, J. Cossy, Angew. Chem. Int. Ed. 59 (2020) 18505-18509. DOI:10.1002/anie.202008572 |
| [46] |
L.C. Li, C.F. Ni, F. Wang, J. Hu, Nat. Commun. 7 (2016) 13320-13331. DOI:10.1038/ncomms13320 |
| [47] |
K. Sekine, A. Ushiyama, Y. Endo, K. Mikami, J. Org. Chem. 85 (2020) 7916-7924. DOI:10.1021/acs.joc.0c00622 |
| [48] |
X. Wang, F. Wang, F.F. Huang, C.F. Ni, J. Hu, Org. Lett. 23 (2021) 1764-1768. DOI:10.1021/acs.orglett.1c00190 |
| [49] |
X.X. Liu, J. Chen, C.Y. Yang, et al., Org. Lett. 23 (2021) 6831-6835. DOI:10.1021/acs.orglett.1c02392 |
| [50] |
F. Sladojevich, A. Trabocchi, A. Guarna, D.J. Dixon, J. Am. Chem. Soc. 133 (2011) 1710-1713. DOI:10.1021/ja110534g |
| [51] |
J. Liu, Z. Fang, Q. Zhang, Q. Liu, X. Bi, Angew. Chem. Int. Ed. 52 (2013) 6953-6957. DOI:10.1002/anie.201302024 |
| [52] |
M. Gao, C. He, H. Chen, et al., Angew. Chem. Int. Ed. 52 (2013) 6958-6961. DOI:10.1002/anie.201302604 |
| [53] |
J.Y. Liao, P.L. Shao, Y. Zhao, J. Am. Chem. Soc. 137 (2015) 628-631. DOI:10.1021/ja511895q |
| [54] |
X.L. He, H.R. Zhao, X. Song, et al., ACS Catal. 9 (2019) 4374-4381. DOI:10.1021/acscatal.9b00767 |
| [55] |
Q. Wan, J.H. Xie, C. Zheng, Y.F. Yuan, S.L. You, Angew. Chem. Int. Ed. 60 (2021) 19730-19734. DOI:10.1002/anie.202107767 |
| [56] |
J.M. Longmire, B. Wang, X.M. Zhang, J. Am. Chem. Soc. 124 (2002) 13400-13401. DOI:10.1021/ja025969x |
| [57] |
C. Chen, X.D. Li, S.L. Schreiber, J. Am. Chem. Soc. 125 (2003) 10174-10175. DOI:10.1021/ja036558z |
| [58] |
X.F. Bai, Z. Xu, C.G. Xia, Z.J. Zheng, L.W. Xu, ACS Catal. 5 (2015) 6016-6020. DOI:10.1021/acscatal.5b01685 |
| [59] |
Z. Chen, N. Ren, X.X. Ma, et al., ACS Catal. 9 (2019) 4600-4608. DOI:10.1021/acscatal.9b00846 |
| [60] |
L. Wei, X. Chang, C.J. Wang, Acc. Chem. Res. 53 (2020) 1084-1100. DOI:10.1021/acs.accounts.0c00113 |
| [61] |
G.F. Zha, W.Y. Fang, J. Leng, H.L. Qin, Adv. Synth. Catal. 361 (2019) 2262-2267. DOI:10.1002/adsc.201900104 |
| [62] |
T. Guo, X.N. Wei, M. Zhang, et al., Chem. Commun. 56 (2020) 5751-5754. DOI:10.1039/d0cc00043d |