 2020, Vol. 31
2020, Vol. 31 


b The Center of New Energy Materials and Technology, School of Materials Science and Engineering, Southwest Petroleum University, Chengdu 610500, China;
c Engineering Technology Research Center of Henan Province for Solar Catalysis, Collaborative Innovation Center of Water Security for Water Source Region of Mid-Line of South-to-North Diversion Project of Henan Province, Nanyang Normal University, Nanyang 473061, China
Methane (CH4) is widely used as an essential fuel and chemical feedstock [1, 2]. The release of CH4 during its storage, transportation, and use has long been neglected, which makes a significant contribution to its concentration increases in the atmosphere. In recent decades, various relevant researches pointed out the greenhouse effect of CH4 is twenty times greater than that of the effect of carbon dioxide [3, 4]. Therefore, decreasing the CH4 emission is highly demanded to migrate the worldwide global warming caused by the greenhouse effect. The studies on effective utilization methods for CH4 conversion have been paid more and more attention [5]. Among various methods for CH4 conversion, steam reforming of CH4 (SRM: CH4 + H2O → 3H2 + CO) to produce hydrogen and syngas over heterogenous catalysts showed a promising way to utilization of CH4 [6-9]. However, CH4 as a symmetric molecule with an extremely high C—H energy (434 kJ/mol) and a non-polar feature is very stable, a high temperature (normally higher than 1073 K) is essential for SRM. This disadvantage significantly inhibits the development and applications of SRM [10].
Photocatalysis using semiconductors and sunlight has shown great potential in promoting difficult reactions that cannot be occurred under mild conditions [11-15]. Among them, photocatalytic steam reforming of CH4 (PSRM) is exceptionally challenging. This reaction not only demands semiconductors featuring high photocatalytic oxidative activity to break the C—H bond of CH4, but also requires that semiconductors have strong reductive abilities for water splitting. Therefore, it is necessary to explore wide bandgap semiconductors with strong redox potentials for PSRM [16, 17]. In addition, the poor adsorption of reactant gases and the utilization of light energy should also be taken into considerations in the gas-solid-phase photocatalytic reaction [18-21].
Perovskite oxides (ABO3) with flexible chemical composition, photostability, and low cost are promising candidates for PSRM [17]. For example, strontium titanate (SrTiO3) as one of perovskite oxide photocatalysts with a wide bandgap was reported to show moderate photocatalytic activity for CH4 oxidation in the gas phase [22]. However, the efficiency of PSRM over SrTiO3 remains relatively low due to the inadequate utilization of light energy and its poor adsorption of CH4. Cocatalysts such as noble metals and carbon dots are widely used for improving the photocatalytic efficiency of PSRM. Au, Pt, Rh and Pd showed well photocatalytic activity for PSRM [23]. For the case of carbon dots and other carbon contain cocatalysts, it might be not well to use in this stage. Because the recent formation of products is extremely low for the PSRM (a few nmol/h levels), if carbon-contain materials are used, it is difficult to confirm the origin of products without using isotopic labeling experiments. Some noble-metal loaded perovskite oxides such as Rh-K2Ti6O13 [24, 25], Pt-CaTiO3 [26, 27], La-NaTaO3 [28] have been developed for PSRM reaction and exhibited improved photocatalytic activity. The noble metals were reported to efficiently promote photogenerated carriers transfer and light adsorption. For instance, Iwashina et al. have demonstrated that Rh doping could enhance the visible-light absorption of SrTiO3, resulting in the improvement of photocatalytic activity [29]. Moreover, various studies have shown that noble metals can improve the adsorption of gas due to their unique electronic structures [30, 31]. Among these noble-metals, Ag was found to be active for CH4 activation in both thermocatalysis and photocatalysis. The silver cationic species (Ag+) can promote the dissociation of CH4 to form Ag-H bonds [3, 32].Nevertheless, the instability of Ag+ under light irradiation significantly limits its photocatalytic application. Coexisting with metallic silver (Ag0) has been proved to be an effective solution to this issue [33].
In this work, we fabricated Ag0/Ag+ decorated SrTiO3 catalysts through a photodeposition method and used for PSRM. TiO2 was synthesized via a hydrothermal method according to the previous works [34, 35]. Then, 199.7 mg (0.25 mmol) of the as-prepared anatase TiO2 and 664.4 mg (0.25 mmol) Sr(OH)2·8H2O were added into a 15 mL solvent mixture (13 mL of deionized water and 2 mL of ethylene glycol (EG)). After the pH was adjusted to 14.0 with a 12 mol/L NaOH aqueous solution, the precursor mixture was added to a 50 mL Teflon-lined vessel and maintained at 160 ℃ for 24 h, afterward cooled down to ambient temperature. Finally, the obtained samples were filtrated and washed by 0.2 mol/L HNO3 solution and deionized water and dried at 60 ℃ for 24 h to obtain SrTiO3 powder.
Ag loaded SrTiO3 was fabricated using a photodeposition method. 200 mg of SrTiO3 was dispersed in 50 mL water by ultrasonic treatment in a round-bottom flask. Then AgNO3 solution (2 mg/mL) and 3 mL isopropanol were added into the suspension. After deoxygenated with Ar gas for 30 min, the flask was illuminated by a Xe lamp (300 W, Perfect light PLS-SXE 300/300UV) for 2 h. The precipitation was centrifugally washed to obtain the solid composites. The catalysts were named as X% Ag-STO, where X represents the molar ratio of Ag in products.
X-ray diffraction (XRD) was carried out on a X'pert diffractometer (PANalytical) with Cu Kα radiation (40 kV, 40 mA). The morphology and microstructure were recorded by transmission electron microscope (TEM) and high resolution transmission electron microscope (HRTEM) (FEI Tecnai G220). X-ray photoelectron spectroscopy (XPS) spectra of catalysts were obtained by X-ray photoelectron analyzer (Thermo ESCALAB 250Xi) and the binding energies were corrected according to the C 1s peak at 284.6 eV. The optical performances were characterized through a UV–vis spectrometer. The N2 adsorption-desorption isotherms were obtained via Quadrasorb SI instrument. Photoluminescence (PL) spectrums were determined on the fluorescence spectrometer (Hitachi F-7000). Surface photovoltage (SPV) tests were proceeded by using a monochromatic-light (Omni-λ 3007) and a SR830 lock-in amplifier. Electron spin resonance (ESR) spectra were observed with a JESFA200 spectrometer.
The reaction tests were conducted in a fixed-bed photoreactor with 380 mL capacity under room temperature (25 ℃), the schematic photocatalytic reactor as shown in Fig. S1 (Supporting information). In a typical process, 10 mL deionized water and 100 mg catalyst diffused uniformly on a glass culture dish were added into the stainless-steel beaker. Then, the photoreactor was flowed with 99.9% Ar gas repeatedly 3 times to remove O2 and CO2. Subsequently, 1 mL CH4 was injected into the photoreactor via a micro-syringe. Finally, the 300 W Xe lamp as the light resource was opened up for the PSRM experiment. 1 mL gas was taken out every hour and analyzed using a Techcomp GC7900. The GC was equipped with a TDX-01 column, flame ionization detector (FID), thermal conductivity detector (TCD), and a CH4 conversion oven. Additionally, in order to verify that the oxidation products are indeed derived from the PSRM reaction, the isotope gas of 13CH4 (m/z 17) was used as the reaction gas to replace 12CH4 (m/z 16) with the detection of products using a mass spectrometer. Photoelectrochemical measurements (PEC) were tested by a CHI 660 workstation. The density functional theory (DFT) calculations were performed using the DMol3 procedure of Materials Studio software [36-38], details of the calculation parameters were shown in the Supporting information.
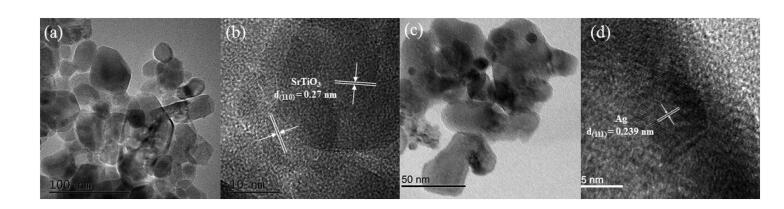
The XRD characterized the composition and crystal structure of X% Ag-STO (X = 0, 0.5 and 1) catalysts. As shown in Fig. S2 (Supporting information), the peaks of 22.8°, 32.4°, 39.9°, 46.5°, 52.6°, 57.8° and 67.8° are indexed as the (100), (110), (111), (200), (210), (211) and (220) planes of cubic SrTiO3 (JCPDS file No. 35-0734) [39]. Compared with pristine SrTiO3, no peak shift was found in Ag-STO, suggesting that Ag was not doped into the lattice structure of SrTiO3 [40]. In addition, no diffraction peak of Ag was observed, which might be due to the small amount and highly dispersed of Ag elements [41-43]. The morphology characteristics of Ag-STO catalysts were studied using TEM and HRTEM (Fig. 1). The nanoparticle distributions of catalysts are similar before and after Ag loading as shown in Figs. 1a and c. The image in Fig. 1b exhibits continuous and well-resolved lattice fringes, with d-spacing of ca. 0.27 nm corresponding to the (110) planes of SrTiO3 [44]. The lattice spacing of 0.239 nm matches well with the (111) crystal planes of Ag (Fig. 1d). These results indicated that the loading of Ag showed no obvious effect on the morphology, shape, size of SrTiO3 catalysts and Ag had been decorated onto the surface of SrTiO3 successfully.

|
Download:
|
| Fig. 1. TEM and HRTEM images of pristine STO (a, b) and 0.5% Ag-STO (c, d). | |
{kind=link}
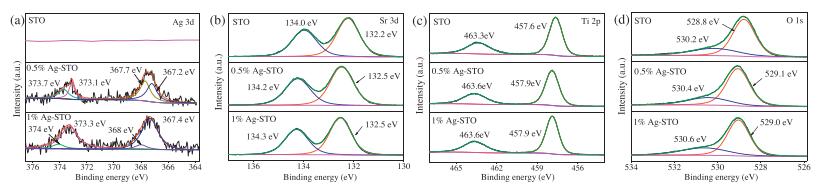
To confirm the state of Ag and other elements on the surface of SrTiO3, the chemical compositions and states of the as-prepared catalysts were revealed by the results of XPS. Fig. S3 (Supporting information) gives the survey spectra of SrTiO3, 0.5% and 1% Ag-STO catalysts. It is difficult to find the peaks of the Ag element in the full-spectra of Ag-STO due to the tiny content of Ag, which is in good agreement with XRD results. However, the high-resolution XPS results clearly showed the existence of Ag element. Each Ag 3d could be further fitted at the binding energy of 373.7/373.1 eV and 367.7/367.2 eV, respectively (Fig. 2a). The peaks at 373.7 and 367.7 eV are assigned to be Ag+ species, and 373.1 and 367.2 eV are assigned to be metallic Ag0 [45]. The atom ratio of Ag+ species to metallic Ag0 on the surface of 0.5% Ag-STO nanocomposites is 51:49. However, when the molar ratio amount of Ag increased to 1%, the atom ratio of Ag+ to Ag0 was changed to 11:89. Fig. 2b shows Sr 3d XPS spectra, the peaks are attributed to Sr2+ state [46]. Ti4+ state is also assigned by 457.6/463.3 eV peaks (Fig. 2c). Moreover, the high-resolution O 1s spectra reveal two peaks, the stronger peak at 528.8 eV is fitted as O2– ions, while another peak is assigned to the surface adsorbed oxygen and hydroxyl groups such as Ti OH species (Fig. 2d) [47]. Compared to the pristine SrTiO3, the peaks of Sr 3d, Ti 2p, O 1s of Ag-STO catalysts all slightly shifted to higher binding energies, this might be due to the strong interactions between Ag and SrTiO3, led to alternate charge transfer in the composite. The XPS result suggested that Ag was successfully loaded onto SrTiO3 and the chemical state of Ag could be changed with the change of Ag content.

|
Download:
|
| Fig. 2. (a) Ag 3d, (b) Sr 3d, (c) Ti 2p, and (d) O 1s XPS spectra of X% Ag-STO. | |
{kind=link}
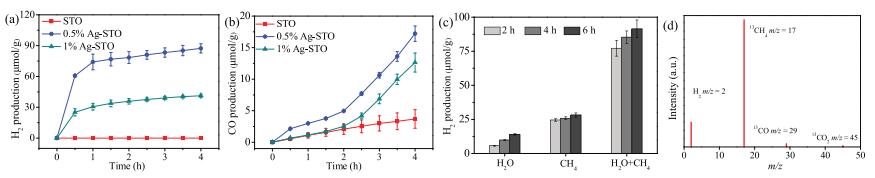
The photocatalytic performances of PSRM over the as-prepared catalysts were estimated under full-spectrum irradiation. The blank experimental results are shown in Fig. S4 (Supporting information). No substance could be observed in the absence of light irradiation or photocatalyst. The PSRM data were calibrated according to the blank test results. The photocatalytic performance of Ag-STO (0.5%) exceeded all other catalysts with a maximum H2 evolution rate of 21.8 μmol g-1 h-1, and a CO production rate of 4.3 μmol g-1 h-1 which is 4.7 times higher than that of SrTiO3 (Figs. 3a and b). Fig. S5 (Supporting information) shows the changing trend of CO2 formation. The trend of CO2 formation is consistent with that of H2 and CO, whereas 0.5% Ag-STO sample showed the highest formation of CO2 with a mount of 10 μmol/g after 4 h irradiation. Besides, the generation of H2 and CO had distinctly different variation trend, the photoirradiated side of catalyst turned brown. These phenomena could be attributed to the formation of medium products and/or carbon deposition in a close system. Yoshida et al. proposed [CH2O]n, ad might be one of the possible expressions as a composition formula, and the formation of [CH2O]n, ad can be depicted as follows (Eq. 1) [16].
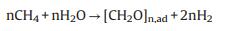
|
(1) |
With the reaction going in the close system, the inactive organic surface species were formed [16, 26], the rate of H2 production decreased while the active reaction intermediates could be persistently decomposed into water and/or carbonaceous com-pound such as CO and CO2. In order to investigate the stability of Ag, the XPS spectrum of 0.5% Ag-STO after reaction was conducted (Fig. S6 in Supporting information). The result confirms that Ag0 and Ag+ can exist stably in the present reaction process. In the presence of a gas mixture of H2O and CH4, the H2 production was the highest, indicating that H2 can be derived from both CH4 and H2O (Fig. 3c). Fig. 3d shows the result of l3C isotope experiment, it is clear that the oxidation products of CO (m/z 29) and CO2 (m/z 45)] contain l3C. Although mass spectrometry is a semi-quantitative analysis method, it can be seen from the signals of l3CO and l3CO2 that l3CO has bigger relative amount, indicating that the 0.5% Ag-STO has a better selectivity for CO production. These results verified the photocatalytic activity of SrTiO3 can be significantly improved through coupling with Ag.

|
Download:
|
| Fig. 3. (a, b) The photocatalytic activities of samples. (c) H2 generation in the presence of only CH4, only H2O and the mixture of CH4 and H2O and (d) 13CH4 isotope calibration results of 0.5% Ag-STO. | |
{kind=link}
To understand the enhanced PSRM efficiency of Ag-STO, the following three characteristics of catalysts were analyzed: reactant gas adsorption, solar light absorption and charge carriers transfer. The adsorption of reactants is the first step in a typical gas-solid-phase photocatalytic process, which has an important influence on photocatalytic activity. It is well known that the adsorption performance is closely related to the surface area and pore structure of catalysts. Fig. S7 (Supporting information) and Table S1 (Supporting information) show the CH4 adsorption-desorption capacity, BET surface area and pore structure of X% Ag-STO, the obtained 0.5% Ag-STO possesses the highest CH4 adsorption capacity and the changing trend of CH4 desorption curves over samples is consistent with that of CH4 adsorption. The BET surface area and pore structure results revealed that there is no significant influence of the BET and pore structure on the CH4 adsorption-desorption capacity of the photocatalyst. Therefore, the addition of Ag elements should be responsible for the enhancement of the CH4 adsorption-desorption.
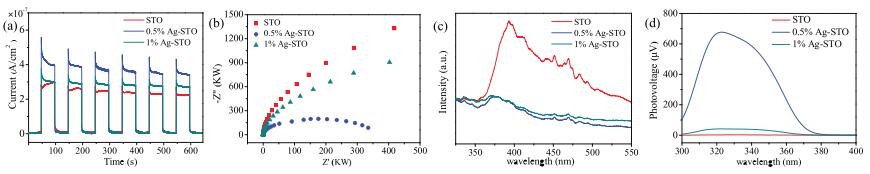
Then the light absorption properties of X% Ag-STO were measured. As shown in Fig. S8 (Supporting information), an additional absorption peak around 400 600 nm was observed for Ag-STO, attributed to the characteristic surface plasmon resonance (SPR) effect of metallic Ag [41, 42, 48]. The deposition of Ag could improve light absorption, resulting in more efficiently photo-generated charge carriers [49, 50]. However, the increase of catalyst absorbance did not lead to enhanced PSRM activity. Therefore, the characteristics of photogenerated charge carriers should be taken into consideration. As shown in Figs. 4a and b, the Ag-STO catalysts showed higher photocurrents and smaller electrochemical impedance spectroscopy (EIS) curves than that of bare STO, However, when the Ag content increased to 1.0 mol%, the photocurrent peak decreases and the diameter of EIS curves increases. The results indicated that Ag can improve the charge carrier transfer, whereas the promoting effect becomes weaker at high loading ratio due to the fact that Ag also could be the hole-electron recombination sites [50, 51]. The PL intensity and SPV patterns of the catalysts are shown in Figs. 4c and d. 1% Ag-STO and 0.5% Ag-STO possess a lower PL intensity and stronger photovoltage as compared to that of bare SrTiO3, which further confirms that the coexistence of a suitable amount of Ag0 and Ag+ can synergistically promote charge separation and lead to higher charge mobility [41, 52, 53].

|
Download:
|
| Fig. 4. (a) Transient photocurrent response, (b) EIS, (c) PL and (d) SPV patterns of the prepared samples. (The photoelectrochemical measurements over the obtained samples at open circuit potential were tested under UV–vis light irradiation, the 0.5 mol/L Na2SO3 solution as electrolyte). | |
{kind=link}
To further determine whether Ag0 and/or Ag+ affects the activity of PSRM, a theoretical calculation was performed. Firstly, the CH4 adsorption capacity of the photocatalyst was studied and shown in Fig. S9 (Supporting information). The Eads of CH4 adsorbed on the surface of pure SrTiO3 and Ag0-STO are -0.37 eV and -0.27 eV, respectively. Given the little difference of adsorption energy between those two materials, it indicates that not Ag0 but Ag+ species might promote the adsorption of CH4. The HOMO orbitals of the Ag-STO catalyst are mainly aggregate on Ag+ species rather than lattice O atoms of SrTiO3 (Fig. S10 in Supporting information), indicating that the electron donors could be Ag+ species, so it is possible that the electrons transition happened around Ag+ after light irradiation. Thus, Ag+ could play as the role to active CH4 molecules during the process of photocatalytic reactions. Furthermore, the reaction of H2O on the catalyst surface is also described by DFT. Since the reduction reaction occurs on the surface of Ag0 particles, Ag atom is placed on the computational model. The Eads of H2O on the surface are -0.21 eV and -1.82 eV for the SrTiO3 and Ag-STO, respectively (Figs. S11 in Supporting information). Therefore, the existence of Ag0 could promote the adsorption of H2O. Interestingly, the H atom of H2O directly transfers to the O atom which is nearby the loading Ag0. Then, the O—H bond and dissociative hydroxyl group form. The hydroxyl groups can react with photoinduced holes to generate ·OH radicals which play a vital role to break the C—H bonds of CH4 [54, 55]. To confirm the formation of ·OH, ESR spectra were performed on 0.5% Ag-STO and SrTiO3 catalysts as shown in Fig. S12 (Supporting information). The signals of O2¯ are also measured due to its highly oxidative. According to the previous researches, O2¯ can promote peroxide CH4 and produce CO2 [56, 57]. Besides, both the 0.5% Ag-STO and SrTiO3 catalysts were detected with the unique four-signal peak of ·OH. Even though there is a certain disparity between ESR results and the specific process of PSRM experiments and the ·OH signal intensity of samples are relatively similar, but the changing trend is identical. Therefore, the stronger signal peaks of the 0.5% Ag-STO could indicate that the promoted photocatalytic activity might be related to ·OH participation.
Based on these evidences, the photoreaction of Ag-STO is described as the following steps. Firstly, electron and hole pairs were generated in conductive and valance bands after light irradiation, respectively (Eq. 2), then carriers migrated to the surface of the catalyst. DFTcalculations verified that the Ag+ on the surface of the catalyst was as the electron donor and the lattice Ti was as the receptor of photogenerated electrons in the process of PSRM reaction. H2O molecules can be easily split to H+ and OH by Ag nanoparticles (Eq. 3). After that, the OH- ions were transferred to Ag+ species and reacted with photogenerated holes to form ·OH radicals (Eq. 4) and dehydrogenate CH4 to generate the surface organic intermediates [CH2O]n, ad (Eq. 5)]. Then the reaction intermediates reacted with OH- ions and photogenerated holes to produce CO and H2O (Eq. 6). The adsorbed CO could be further oxidized to form CO2 (Eq. 7). It is generally believed that noble metal could accept photoexcited electrons and play a role in photocatalytic reduction reactions. Consequently, the photoexcited accumulated electrons on the surface of SrTiO3 preferably transferred to Ag0. The protons can be reduced by these electrons on the surface of Ag0 species to form molecular hydrogen (Eq. 8). The photocatalytic mechanism is proposed in Scheme S1 (Supporting information).
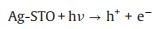
|
(2) |
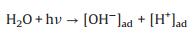
|
(3) |
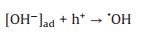
|
(4) |
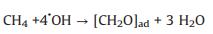
|
(5) |
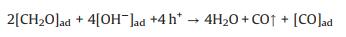
|
(6) |
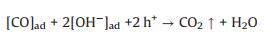
|
(7) |
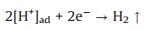
|
(8) |
In summary, PSRM was achieved over Ag-SrTiO3 photocatalysts. The highest activity was acquired over 0.5 mol% Ag-STO prepared by the photodeposition method, where metallic Ag0 and Ag+ species coexisted on the surface of SrTiO3 with a suitable surface ratio. A synergistic effect was achieved to improve the photocatalytic performance. The Ag0 can not only extend the light absorption range and be regarded as the reduction reaction sites during the process of PSRM, but also can significantly promote the adsorption of H2O to produce ·OH radicals. While the Ag+ species can enhance the adsorption of CH4 and be regarded as the oxidation reaction sites. Those two different valence states of Ag functioned together to promote the separation of electron-holes. The coexisted Ag0 and Ag+ effect with a suitable ratio was found to improve the photocatalytic activity for SRM. The highest CO production rate of 4.3 μmol g-1 h-1 can be obtained, which is approximately 5 times higher than that of pure SrTiO3. Finally, the specific and deeper investigation of the PSRM reaction process should lead to the development of efficient photocatalysts.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge the financial support from the Sichuan Provincial International Cooperation Project (Nos. 2017HH0030, 2019YFH0164), the National Natural Science Foundation of China (No. 21403172). We appreciate the calculation support from the National Supercomputing Center in Shenzhen.
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.11.007.
| [1] |
L. Li, S.Z. Fan, X.Y. Mu, Z.T. Mi, C.J. Li, J. Am. Chem. Soc. 136 (2014) 7793-7796. |
| [2] |
J.H. Chen, H. Arandiyan, X. Gao, J.H. Li, Catal. Surv. Asia 19 (2015) 140-171. DOI:10.1007/s10563-015-9191-5 |
| [3] |
X.X. Chen, Y.P. Li, X.Y. Pan, et al., Nat. Commun. 7 (2016) 12273.. DOI:10.1038/ncomms12273 |
| [4] |
S. Kirschke, P. Bousquet, P. Ciais, M. Saunois, G. Zeng, Nat. Geosci. 6 (2013) 813-823. DOI:10.1038/ngeo1955 |
| [5] |
S. Murcia-Lo'pez, K. Villa, T. Andreu, J.R. Morante, ACS Catal. 4 (2014) 3013-3019. DOI:10.1021/cs500821r |
| [6] |
J.N. Armor, Appl. Catal. A:Gen. 176 (1999) 159-176. DOI:10.1016/S0926-860X(98)00244-0 |
| [7] |
J.L.G. Fierro, M.A. Pena, J.P. Gomez, Appl. Catal. A:Gen. 144 (1996) 7-57. DOI:10.1016/0926-860X(96)00108-1 |
| [8] |
J.R. Rostrupnielsen, Catal. Today 18 (1993) 305-324. DOI:10.1016/0920-5861(93)80059-A |
| [9] |
K. Shimura, H. Yoshida, Catal. Surv. Asia 18 (2014) 24-33. DOI:10.1007/s10563-014-9165-z |
| [10] |
A. Amin, A. Abedi, R. Hayes, M. Votsmeier, W. Epling, Appl. Catal. A:Gen. 478 (2014) 91-97. DOI:10.1016/j.apcata.2014.03.032 |
| [11] |
Y. Shan, Y.Q. Zhong, B.Q. Yu, S.Y. Cai, Y. Zhou, Phys. Chem. Chem. Phys. 18 (2016) 20338-20344. DOI:10.1039/C6CP02561G |
| [12] |
D. Wang, X.Y. Pan, G.T. Wang, Z.G. Yi, J. Struct. Chem. 37 (2018) 230-241. |
| [13] |
Y.Z. Cai, X.Y. Pan, Y.P. Li, Z.G. Yi, J. Struct. Chem. 35 (2016) 1328-1338. |
| [14] |
K. Tamai, K. Murakami, S. Hosokawa, et al., J. Phys. Chem. C 121 (2017) 22854-22861. DOI:10.1021/acs.jpcc.7b07339 |
| [15] |
R. Abe, H. Takami, N. Murakami, B. Ohtani, J. Am. Chem. Soc. 130 (2008) 7780-7781. DOI:10.1021/ja800835q |
| [16] |
H. Yoshida, K. Hirao, J. Nishimoto, et al., J. Phys. Chem. C 112 (2008) 5542-5551. |
| [17] |
H. Yoshida, S. Kato, K. Hirao, J. Nishimoto, T. Hattori, Chem. Lett. 36 (2007) 430-431. DOI:10.1246/cl.2007.430 |
| [18] |
S. Delavari, N.A.S. Amin, M. Ghaedi, J. Clean. Prod. 111 (2015) 143-154. |
| [19] |
J.F. Muir, R.E. Hogan Jr., R.D. Skocypec, R. Buck, Sol. Energy 52 (1990) 467-477. |
| [20] |
Y. Li, Y. Cai, X. Chen, et al., RSC Adv. 6 (2016) 2760-2767. DOI:10.1039/C5RA22459D |
| [21] |
L. Li, G.D. Li, C. Yan, et al., Angew. Chem. Int. Ed. 50 (2011) 8299-8303. DOI:10.1002/anie.201102320 |
| [22] |
X.Y. Pan, X.X. Chen, Z.G. Yi, Phys. Chem. Chem. Phys. 40 (2016) 163-181. |
| [23] |
K. Shimura, T. Yoshida, H. Yoshida, J. Phys. Chem. C 114 (2010) 11466-11474. DOI:10.1021/jp1012126 |
| [24] |
K. Shimura, H. Kawai, T. Yoshida, H. Yoshida, Chem. Commun. (Camb.) 47 (2011) 8958-8960. DOI:10.1039/c1cc12287h |
| [25] |
K. Shimura, H. Kawai, T. Yoshida, H. Yoshida, ACS Catal. 2 (2012) 2126-2134. DOI:10.1021/cs2006229 |
| [26] |
K. Shimura, H. Yoshida, Energy Environ. Sci. 3 (2010) 615-617. DOI:10.1039/b922793h |
| [27] |
K. Shimura, H. Miyanaga, H. Yoshida, Stud. Surf. Sci. Catal. 175 (2010) 85-92. DOI:10.1016/S0167-2991(10)75011-4 |
| [28] |
K. Shimura, S. Kato, T. Yoshida, et al., J. Phys. Chem. C 114 (2010) 3493-3503. DOI:10.1021/jp902761x |
| [29] |
K. Iwashina, A.J.J. Kudo, J. Am. Chem. Soc. 133 (2011) 13272-13275. DOI:10.1021/ja2050315 |
| [30] |
H.J. Freund, M.W. Roberts, Surf. Sci. Rep. 25 (1996) 225-273. DOI:10.1016/S0167-5729(96)00007-6 |
| [31] |
R.Y. Zhang, Z.A. Huang, C.G. Li, Y.S. Zuo, Y. Zhou, Appl. Surf. Sci. 475 (2019) 953-960. DOI:10.1016/j.apsusc.2019.01.050 |
| [32] |
T. Baba, H. Sawada, T. Takahashi, M. Abe, Appl. Catal. A:Gen. 231 (2002) 55-63. DOI:10.1016/S0926-860X(01)00904-8 |
| [33] |
X.F. Wang, S.F. Li, H.G. Yu, J.G. Yu, S.W. Liu, Chem.Eur. J. 17 (2011) 7777-7780. DOI:10.1002/chem.201101032 |
| [34] |
W.W. Chen, S. Yu, Y.Q. Zhong, et al., New J. Chem. 42 (2018) 4811-4817. DOI:10.1039/C8NJ00180D |
| [35] |
S.Y. Cai, S. Yu, W.C. Wan, W. Wen, Y. Zhou, RSC Adv. 7 (2017) 27397-27404. DOI:10.1039/C7RA02433A |
| [36] |
Y.X. Yang, M.G. White, P. Liu, J. Phy. Chem. A 116 (2011) 248-256. |
| [37] |
J.P. Perdew, K. Burke, M. Ernzerhof, Phy. Rev. Lett. 77 (1996) 3865-3868. DOI:10.1103/PhysRevLett.77.3865 |
| [38] |
F. Wang, Y.H. Ye, Y.H. Cao, Y. Zhou, Appl. Surf. Sci. 481 (2019) 604-610. DOI:10.1016/j.apsusc.2019.03.079 |
| [39] |
X.Y. Wu, X.Y. Wang, J. Li, G. Zhang, J. Mater. Chem. A 5 (2017) 23822-23830. DOI:10.1039/C7TA08061A |
| [40] |
P. Aprelev, Y. Gu, R. Burtovyy, I. Luzinov, K.G. Kornev, J. Appl. Phys. 118 (2015) 3806-3812. |
| [41] |
Q. Zhang, Y. Huang, L.F. Xu, et al., ACS Appl. Mater. Interfaces 6 (2016) 4165-4172. |
| [42] |
Y. Yu, Z.M. Lu, Q. Guo, et al., Chem. Phys. Lett. 692 (2018) 94-101. DOI:10.1016/j.cplett.2017.12.001 |
| [43] |
B.W. Ma, J.F. Guo, W.L. Dai, K.G. Fan, Appl. Catal. B:Environ. 123- 124 (2012) 193-199. |
| [44] |
S.X. Ouyang, P. Li, H. Xu, et al., ACSAppl.Mater.Interfaces 6 (2014) 22726-22732. DOI:10.1021/am506877b |
| [45] |
Y.X. Yang, W. Guo, Y.N. Guo, et al., J. Hazard. Mater. 271 (2014) 150-159. DOI:10.1016/j.jhazmat.2014.02.023 |
| [46] |
Y.Q. Bi, M.F. Ehsan, Y. Huang, J.R. Jin, T. He, J. CO2 Util. 12 (2015) 43-48. DOI:10.1016/j.jcou.2015.10.004 |
| [47] |
Q. Zhang, Y. Huang, S.Q. Peng, et al., Appl. Catal. B:Environ. 239 (2018) 1-9. DOI:10.1016/j.apcatb.2018.07.076 |
| [48] |
Z. Zhang, Y. Zhou, S. Yu, M.L. Chen, F. Wang, Mater. Lett. 150 (2015) 97-100. DOI:10.1016/j.matlet.2015.03.011 |
| [49] |
Y. Zhou, Q. Zhang, Y.H. Lin, et al., Sci. China Chem. 56 (2013) 435-442. DOI:10.1007/s11426-013-4846-4 |
| [50] |
F. Dong, Q.Y. Li, Y. Zhou, et al., Dalton Trans. 43 (2014) 9468-9480. DOI:10.1039/C4DT00427B |
| [51] |
L. Jin, G.Q. Zhu, M. Hojamberdiev, X.C. Luo, P. Liu, Ind. Eng. Chem. Res. 53 (2014) 2127-2134. |
| [52] |
A. Lebedev, F. Anariba, X. Li, D. Seng Hwee Leng, P. Wu, Sol. Energy 178 (2019) 257-267. DOI:10.1016/j.solener.2018.12.040 |
| [53] |
F. Chen, H.W. Huang, Y.H. Zhang, T.R. Zhang, Chin. Chem. Lett. 28 (2017) 2244-2250. DOI:10.1016/j.cclet.2017.09.017 |
| [54] |
L.H. Yu, Y. Shao, D.Z. Li, Appl. Catal. B:Environ. 204 (2017) 216-223. DOI:10.1016/j.apcatb.2016.11.039 |
| [55] |
K. Villa, S. Murcia-López, T. Andreu, J.R. Morante, Appl. Catal. B:Environ. 163 (2015) 150-155. DOI:10.1016/j.apcatb.2014.07.055 |
| [56] |
M.A. Gondal, A. Hameed, A. Suwaiyan, Appl. Catal. A:Gen. 243 (2003) 165-174. DOI:10.1016/S0926-860X(02)00562-8 |
| [57] |
G.R. Bamwenda, T. Uesigi, Y. Abe, K. Sayama, H. Arkawa, Appl. Catal. A:Gen. 205 (2001) 117-128. DOI:10.1016/S0926-860X(00)00549-4 |