 2018, Vol. 29
2018, Vol. 29 



b Laboratory of Inorganic Synthesis and Bioinspired Catalysis(LISBIC), Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India
Iron-sulfur carbonyls such as [(μ-S)2Fe2(CO)6] [1], [(μ-SR)2 Fe2(CO)6] [2] and [(μ-S)(μ-SR)Fe2(CO)6] [3] are very known in organometallic chemistry, and bioinorganic chemistry as well [4]. In particular, the active site of [FeFe]-H2ase, also known as H-cluster, is composed of a {2Fe2S} core that is attached by a redoxactive [Fe4S4] cluster [5]. As a family of bimetallic hydrogenase, it catalyzes the interconversion of H2, electrons and protons at remarkably fast rate and with a low over potential [6]. In {2Fe2S} core, the diiron centers are assembled by an azadithiolate ligand (SCH2NHCH2S-) [7] and a CO bridging ligand, while each iron center is coordinated by one CO and one CN- ligand [8]. Overall, the active site can be formulated as [(Cys-S-Fe4S4)(NC)(OC)Fe {(SCH2)2NH}Fe(CO)2(CN)], and the key structural feature is the rotated Fed center that exhibits a vacant coordination site trans to the bridging CO for binding dihydrogen or a hydride ligand during the catalysis (Fig. 1a). Since the reports of the crystallographic structure of the active site in the late of 1990s, intensive efforts have been devoted to the synthetic models of [FeFe]-H2ase aiming at exploring efficient iron-based catalysts for H2 production [9]. The modeling efforts have focused on the substitution CO ligands of parent complex [(μ-SR)2 Fe2(CO)6] by a variety of more basic ligands such as phosphines [10], cyanide [11], N-heterocyclic carbenes [12], providing synthetic models of [(μ-SR)2 Fe2(CO)6-xLx] type (Fig. 1b).

|
Download:
|
| Fig. 1. (a) Active site structure of [FeFe] -H2ases; (b) simplified structure of models relative to the active site of [FeFe] -H2ases. | |
{kind=link}
The phosphine ligands have been found to exhibit a range of steric and electronic effects to the diiron center in [(μ-SR)2 Fe2(CO)6-x(PR3)x] class. For example, symmetrical bisPMe3 compound [Fe2(pdt)(CO)4(PMe3)2] (pdt = CH2(CH2S-)2) allows its Fe(Ⅰ)-Fe(Ⅰ) bond to undergo protonation giving the bridging hydride [(μ-H)Fe2(pdt)(CO)4(PMe3)2]+ [13]. The cationic diferrous bridging hydrides [Fe(Ⅱ)HFe(Ⅱ)]+ can be reduced to the neutral mixed-valence Fe(Ⅰ)HFe(Ⅱ) hydrides, which are susceptible to protonation resulting in H2 evolution [14]. Protonating unsymmetrically substituted compounds such as [(CO)3Fe(pdt)-Fe (dppv)(CO)] (dppv = cis-1, 2-C2H2(PPh2)2) bearing a chelating diphosphine ligand at one of the iron centers gives terminal hydrides [(t-H)Fe(Ⅱ)Fe(Ⅱ)]+ at low temperatures [15]. More likely as a kinetic product, the terminal hydrides transform to the thermodynamically stable bridging hydride [(μ-H)Fe(Ⅱ)Fe(Ⅱ)]+. The life time of the terminal hydride species can be prolonged by the increase in steric bulk of the phosphine ligands [16], and even be stabilized by the introduction of an anionic ligand C5Me5-, i.e., [(C5Me5)Fe(pdt)Fe(CO)(dppe)H] [17]. Synthesis of [Fe(Ⅰ)Fe(Ⅰ)] diiron compounds with the rotated structure is challenging. The possibilities have been explored by using bulky dithiolate ligands with the combination of unsymmetrical ligand substitution [18]. Models of [Fe(Ⅰ)Fe(Ⅱ)] with a rotated configuration have been reported by one-electron oxidation of the unsymmetrical substituted Fe(Ⅰ)Fe(Ⅰ) precursors such as [(IMes)(CO)2Fe(SC3H6S)Fe (PMe3)(CO)2] (IMes = 1, 3-bis(2, 4, 6-trimethylphenyl)imidazol-2- ylidene) [19], and [(PMe3)(CO)2Fe(SC2H4S)Fe(CO)2(dppv)] [20]. The reactivity of [Fe(Ⅰ)Fe(Ⅱ)]+ models is indeed interesting, because it has been found that in the presence of base oxidizing [Fe(Ⅰ)Fe(Ⅱ)]+ models enables H2 splitting [21]. Especially, the CO ligands in [Fe(Ⅰ)Fe(Ⅱ)]+ compounds can be replaced by more basic ligands such as phosphines, providing new synthetic complexes for further investigation [22].
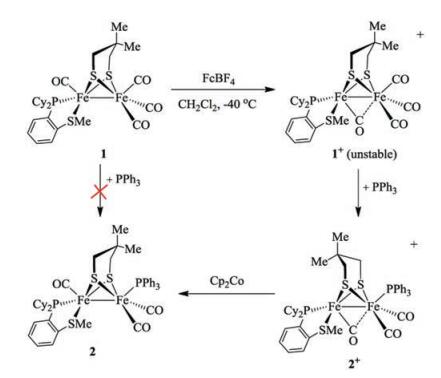
Although the {2Fe2S} core of the active site contains a sulfur ligated Fe center (Fed, Fig. 1a), synthetic {2Fe3S} models with a thioether ligand have been rarely reported [23]. In this paper, we report a series of {2Fe3S} complexes bearing phosphino thioether chelating ligand (1, 2-Cy2PC6H4SMe) based on [Fe2(Me2pdt)(1, 2-Cy2PC6H4SMe)(CO)4] (Me2pdt = Me2C(CH2S-)2, 1). Compound 1 exhibits reversible one-electron redox even for [Fe(Ⅰ)Fe(Ⅱ)]+/0 couple. The synthesis of tri-substituted [Fe(Ⅰ)Fe(Ⅱ)]+ cationic complex [Fe2(Me2pdt)(1, 2-Cy2PC6H4SMe)(PPh3)(CO)3]+ ([2]+) was achieved by installing a phosphine ligand on the in-situ oxidized form of the [Fe(Ⅰ)Fe(Ⅰ)] precursors (Scheme 1). Reduction of [2]+ provides the neutral tri-substituted compound 2. The reactivity of 1 and 2 towards different strength of acids were investigated.

|
Download:
|
| Scheme 1. Synthetic route for {2Fe3S} complexes bearing a phosphino thioether and chelating ligand. | |
{kind=link}
We initially attempted to install 1, 2-Cy2PC6H4SMe on the parent compound [Fe2(Me2pdt)(CO)6] by nucleophilic abstraction of carbonyl using Me3NO [24]. However, this reaction resulted in a mixture of 1 and the undesired mono-substituted product [Fe2(Me2pdt)(1, 2-Cy2PC6H4SMe)(CO)5]. When 1, 2-Ph2PC6H4-SMe was used as the ligand, the mono-substituted product [Fe2(Me2pdt)(1, 2-Ph2PC6H4SMe)(CO)5] was solely produced (Fig. S16 in Supporting information).
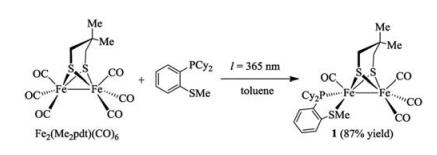
Alternatively, we synthesized 1 by irradiation (λ = 365 nm) of a mixture of [Fe2(Me2pdt)(CO)6] and 1, 2-Cy2PC6H4SMe (1:1 ratio) in toluene (Scheme 2). After four hours of photolysis, FI-IR spectroscopy indicated complete conversion of [Fe2(Me2pdt)- (CO)6] to the product. After evaporating the solvent, the compounds were readily purified by recrystallization from CH2Cl2 by layering hexane and isolated as brown solids (ca. 87% yields). The IR spectrum of 1 displays its νCO bands at 2010, 1939, and 1894 cm-1, which is ~10 cm-1 lower in energy compared to [Fe2(pdt)(dppbz)(CO)4] (dppbz = 1, 2-bis(diphenylphosphanyl)benzene) (νCO = 2019, 1949 and 1906 cm-1) [25] and [Fe2(pdt)(dppv)(CO)4] (νCO = 2021, 1950 and 1912 cm-1) [24].

|
Download:
|
| Scheme 2. Synthesis of complex 1. | |
{kind=link}
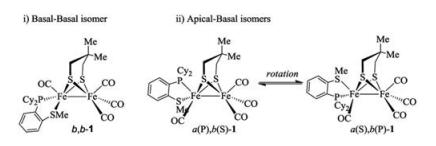
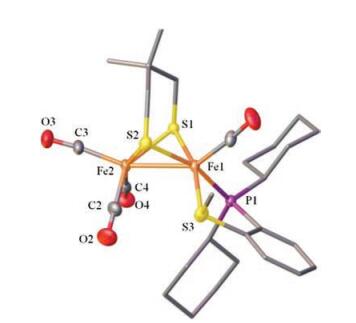
Similar to the reported diphosphine-based compounds [Fe2(pdt)(P-P)(CO)4] (P-P = chelating diphosphine ligand) [18b, 24], complex 1 also exhibit isomers as indicated by 31P NMR spectroscopic studies. At room temperature, the 31P NMR spectrum displays two sets of resonances as one broad peak at δ 90.7 and a singlet at δ 78.2 in a ratio of 1:4. The binding of phosphine atom at [Fe(P-SR)(CO)] subunit can either be apical or basal leading to the existence of at least three isomers as shown in Fig. 2. The broad phosphorus resonances at δ 90.7 are tentatively assigned for the apical-basal isomers, while the sharp singlet is assigned to the basal-basal isomer. The broad phosphorus resonances might arise from the rapid turnstile rotation of the phosphino thioether ligand bound at the iron center in an apicalbasal fashion (Fig. 2). To confirm the existence of such a dynamic movement, we have conducted variable-temperature 31P NMR experiments for 1 (Fig. 3). Upon cooling of the sample from 293 K to 213 K, the signal at δ 90.7 ppm shifted to δ 95.0 ppm, whereas the singlet retained at δ 78.2 ppm. Importantly, variable-temperature 31P NMR spectra showing coalescences and decoalescences suggest the rapid interconversion between a(P), b(S)-1 and a(P), b (S)-1 for the apical-basal isomers. The structure for the basal-basal isomer b, b-1 was confirmed by single crystal X-ray diffraction (Fig. 4).

|
Download:
|
| Fig. 2. Proposed basal-basal (ⅰ) and apical-basal (ⅱ) isomers for 1. | |
{kind=link}

|
Download:
|
| Fig. 3. 31P NMR spectra recorded for 1 in CD2Cl2 at various temperatures from 298 K to 213 K. | |
{kind=link}

|
Download:
|
| Fig. 4. Molecular structures of b, b-1 with 50% thermal ellipsoids. H atoms are omitted for clarity. Selected distances (Å) and angles (deg): Fe1-Fe2 2.5725(5), Fe1-S3 2.2493(7), C1-Fe1 1.742(2), Fe1-Fe2-C3 155.35(7). | |
{kind=link}
In CH2Cl2, cyclic voltammogram of 1 shows a reversible oxidation event at -0.28 V (vs. Fc/Fc+) for the Fe(Ⅰ)Fe(Ⅰ)/Fe(Ⅰ)Fe(Ⅱ) couple (Fig. S18 in Supporting information). The oxidation potential is comparable to -0.34 V for [Fe2(Me2pdt)(PMe3)2(CO)4] [26], and is slightly more negative than -0.19 V for [Fe2(pdt)(dppv) (CO)4] [27]. For the unsymmetrical substituted [Fe(Ⅰ)Fe(Ⅰ)] precursors, one-electron oxidation provides [Fe(Ⅰ)Fe(Ⅱ)]+ cationic complex featuring a bridging or semi-bridging CO ligand [19a, 23b, 26]. The oxidation of 1 was subsequently examined by using FcBF4 oxidant. When the CH2Cl2 solution of 1 was treated with an equivalent of FcBF4, the color of the solution immediately turned to purple from brown. In IR spectra, the new νCO bands were observed at 2079, 2021 and 1890 cm-1. The ΔνCO is ~45 cm-1 higher than that of 1, which is consistent with the one-electron oxidation reaction in the preparation of [Fe(Ⅰ)Fe(Ⅱ)]+ cationic complexes [22d, e]. Due to the semibridging CO, there is a vacant site in apical position of Fe1 (16e-) in [Fe(Ⅱ)Fe(Ⅰ)]+ models [21b]. Different from the dynamic movement of 1, we conclude that the dibasal fashion of [1]+ would not turn to apical-basal fashion in order to keep the rotated square-pyramidal structure. However, the attempts to isolate the [Fe(Ⅰ)Fe(Ⅱ)]+ mixed-valent species of [1]+ was unsuccessful.
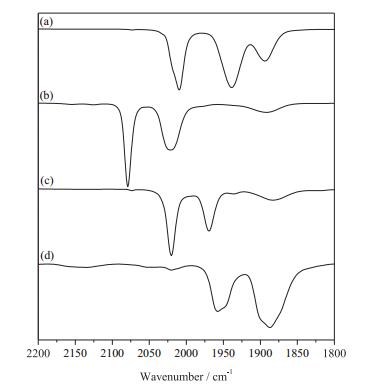
Notably, the [Fe(Ⅰ)Fe(Ⅱ)]+ specie can be stabilized by the introduction of mono-phosphines such as PPh3. To the solution of [1]+ generated in situ from 1 with FcBF4 in CH2Cl2, was added one equivalent of PPh3 affording [2]+. The cationic salts [2]+ was isolated as dark-green solids in 86% yield. Three new νCO bands at 2020, 1989 and 1880 cm-1 were displayed in the IR spectrum (Fig. 5), and the weak broad band at 1880 cm-1 is corresponding to the νμ-CO. The νCO of [2]+ is ~55 cm-1 higher than that of [1]+. Complex [2]BF4 does not undergo further substitution in the presence of excess of PPh3.

|
Download:
|
| Fig. 5. IR spectra (νCO region) for 1 (a, 2009, 1944 and 1907 cm-1), [1]+ generated in situ by 1 with FcBF4 (b, 2079, 2021 and 1890 cm-1), [2]+ from the reaction of [1]+ with PPh3 (c, 2020, 1969 and 1880 cm-1); 2 from the reduction of [1]+ by Cp2Co in CH2Cl2 (d, 1954 and 1887 cm-1). | |
{kind=link}
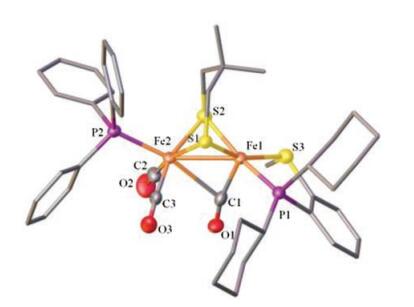
X-ray crystallographic analysis reveals the expected rotated structure for the cationic complex [2]+, which features a semibridging CO ligand (Fig. 6). The Fe(1)-C(1) distance is 1.771(7) Å, and about 0.71 Å shorter than Fe(2)-C(1) (2.480(6) Å). The phosphine ligand PPh3 is bound at apical position of [Fe(CO)2(PPh3)] subunit, while the phosphino thioether ligand chelating at the dibasal position of the other iron center. The angle of Fe2-Fe1-C1 (66.86 (19)°) is much smaller than those in [Fe2(pdt)(dppv)(CO)3(PR3)]+ (R = Me, 73.20(13)°; i-Pr, 78.55(16)°) [22e].

|
Download:
|
| Fig. 6. Molecular structure of [2]+ cation with 50% thermal ellipsoids. H atoms and BF4- are omitted for clarity. Selected distances (Å) and angles (deg): Fe1-Fe2 2.5657 (12), Fe1-C1 1.771(7), Fe2-C1 2.480(6), Fe1-C1-O1 165.0(5). | |
{kind=link}
Surprisingly, cyclic voltammogram of [2]+ indicated that the substitution of the CO ligand in 1 by PPh3 results in an anodic shift of the FeⅡFeⅠ/FeⅠFeⅠ couple of 470mV. The reversible redox event at -0.75 V (ipa/ipc = 0.95, vs. Fc+/Fc) suggests the possibility of bulk chemical reduction and isolation of 2. Treatment of [2]+ with one equivalent of Cp2Co (-1.33 V in CH2Cl2 vs. Fc+/Fc) [28] in CH2Cl2 solution afforded 2, which was isolated as brown solids in high yield. In IR spectrum, the νCO bands were shown as two broad peaks at 1954 (br), 1887 (br) cm-1. The 31P NMR spectrum of 2 consists of two groups of singlets at δ 76.1 (P-SR), 54.9 (PPh3) and δ 87.2 (P-SR), 56.9 (PPh3) at room temperature, indicating the existence of two isomers. In addition, the formation of 2 was not observed by irradiation of 1 with PPh3 in toluene judging from 31P NMR spectra.
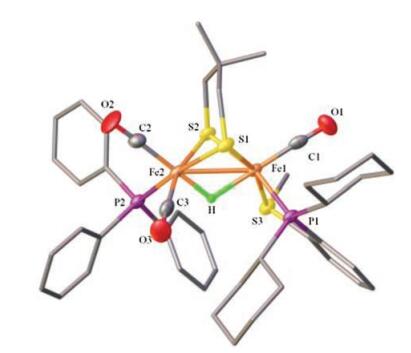
With the addition of HBF4·Et2O to the CH2Cl2 solution of 2, the phosphorous resonances were displayed as two singlets at δ 80.5 for the phosphino thioether ligand (P-SR) and δ 52.7 for PPh3. In the 1H NMR spectrum, the hydride signal was observed at -11.9 ppm (t, JPH = 20 Hz), indicating the formation of bridging hydride complex [H2]BF4. Its structure was confirmed by X-ray crystallographic studies (Fig. 7). In [H2]+, the phosphino thioether ligand retains the dibasal chelating mode while the PPh3 resides at the basal position at [Fe(CO)2(PPh3)] subsite. The PPh3 binding at the basal site dues to the strong trans effect of CO to H- in [H2]+ [29]. The hydride ligand was located in the difference map, and its position was refined. Judging from the bond distances of Fe1-H 1.72 Å and Fe2-H 1.47 Å, the μ-H is semi-bridging.

|
Download:
|
| Fig. 7. Molecular structure of [H2]+ with 50% thermal ellipsoids. Selected bond distances (Å) and angles (deg): Fe1-Fe2 2.6302(15), Fe1-P12.2322(18), Fe2-P2 2.275 (2), Fe1-S3 2.2510(19), Fe1-C1 1.730(8), Fe2-C2 1.769(7), C2-Fe2-Fe1 141.4(2), P1-Fe1-Fe2 114.14(6). | |
{kind=link}
The most striking difference between 1 and 2 is their reactivity towards different strength of acids. Protonating the di-substituted compound 1 requires strong acids such as HBF4·Et2O (pKaMeCN = 0.2) [30] and CF3SO3H (pKaMeCN = 2.6) [31]. When [HPPh3]BF4 (pKaMeCN = 7.6) [32] was used, there was no hydride signal observed in 31P NMR spectrum. By contrast, the tri-substituted compound 2 reacts with CCl2COOH (pKaMeCN = 13.2) [33] producing [H2]+. The pKaMeCN of the Fe-Fe bond in 2 was estimated to be in the range of 13.2–14, because it cannot be protonated by 2, 6-Me2py-H+ (pKaMeCN = 14.2) [34]. The ΔpKaMeCN between 1 and 2 is about 10. It is noteworthy that the tetra-phosphine substituted diiron complex [Fe2(adtNH)-(CO)2(dppv)2] possesses the pKaMeCN in between 15.3 and 17.6 [34].
A series of {2Fe3S} complexes bearing phosphino thioether chelating ligand were synthesized and well characterized. As well as the unsymmetrical substituted [Fe(Ⅰ)Fe(Ⅰ)] compounds with diphosphine chelating ligands, the phosphino-thioether-based disubstituted diiron compound 1 exhibits a reversible one-electron redox event for [Fe(Ⅰ)Fe(Ⅱ)]+/0 couple. The synthesis of trisubstituted [Fe(Ⅰ)Fe(Ⅱ)]+ cationic complex [2]+ was achieved by adding a phosphine ligand to [1]+ generated in situ by the reaction of 1 with FcBF4. Reduction of [2]+ provides the neutral trisubstituted [Fe(Ⅰ)Fe(Ⅰ)] compound 2. The substitution of the CO in 1 ligand by PPh3 results in an anodic shift of the FeⅡFeⅠ/FeⅠFeⅠ couple of 470 mV. Acid-base features of [Fe-Fe] model complexes are important properties [35-37]. Both 1 and 2 undergo protonation affording bridging hydride complexes. Most importantly, this substitution also leads to the Fe-Fe bonds in 1 and 2 with large Lewis basicity difference, i.e. ΔpKaMeCN ~10.
AcknowledgmentsWe gratefully acknowledge the financial support from the "1000 Youth Talents Plan", the Natural Science Foundation of China (Nos. 21402107, 91427303). We also thank Prof. Di Sun for assistance with the X-ray crystallography. S. Raje acknowledges CSIR, India, for a senior research fellow ship. R. Angamuthu thanks IIT Kanpur for funding.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2018.03.013.
| [1] |
(a) Y. Li, T.B. Rauchfuss, Chem. Rev. 116 (2016) 7043-7077; (b) L.C. Song, M.Y. Tang, F.H. Su, et al., Angew. Chem. Int. Ed. 45 (2006) 1130-1133; (c) J.A. Kovacs, J.K. Bashkin, R.H. Holm, J. Am. Chem. Soc.107 (1985) 1784-1786; (d) H. Abul-Futouh, L.R. Almazahreh, M.K. Harb, et al., Inorg. Chem. 56 (2017) 10437-10451. |
| [2] |
(a) R.P. Venkateswara, R.H. Holm, Chem. Rev. 104 (2004) 527; (b) L.C. Song, H. Tan, A.G. Zhu, Y.Y. Hu, H. Chen, Organometallics 34 (2015) 1730-1741; (c) L.C. Song, H.T. Fan, Q.M. Hu, J. Am. Chem. Soc. 124 (2002) 4566-4567; (d) D. Chong, I.P. Georgakaki, M.Y. Darensbourg, et al., Dalton Trans. (2003) 4158-4163. |
| [3] |
(a) L.C. Song, Acc. Chem. Res. 38 (2005) 21-28; (b) L.C. Song, Y.L. Li, Q.M. Hu, et al., Inorg. Chem. 49 (2010) 10174-10182; (c) L.C. Song, L.D. Zhang, B.B. Liu, et al., Organometallics 36 (2017) 1419-1429; (d) S.J. George, Z. Cui, M. Razavet, C.J. Pickett, Chem.-Eur. J. 8 (2002) 4037-4046. |
| [4] |
(a) K. Fauvel, R. Mathieu, R. Poilblanc, Inorg. Chem. 15 (1976) 976-978; (b) U.E. Apfel, F.Y. Pétillon, W. Weigand, et al., [FeFe] Hydrogenase Models: An Overview, in: W. Weigand, P. Schollhammer (Eds.), Bioinspired Catalysis: Metal-Sulfur Complexes, Wiley-VCH Verlag GmbH & Co KGaA, 2014, pp. 79-104; (c) S. Ogo, Y. Mori, M. Asano, et al., Angew. Chem. Int. Ed. 56 (2017) 9723-9726; (d) Y. Shomura, M. Taketa, Y. Higuchi, et al., Science 357 (2017) 928-932; (e) A. Onoda, Y. Kihara, K. Fukumoto, Y. Sano, T. Hayashi, ACS Catal. 4 (2014) 2645-2648. |
| [5] |
E.M. Shepard, E.S. Boyd, J.B. Broderick, J.W. Peters, Curr. Opin. Chem. Biol. 15 (2011) 319-327. |
| [6] |
J.C. Fontecillacamps, A. Volbeda, C. Cavazza, Y. Nicolet, Chem. Rev. 107 (2007) 4273-4303. DOI:10.1021/cr050195z |
| [7] |
A. Silakov, B. Wenk, E. Reijerse, W. Lubitz, Phys. Chem. Chem. Phys. 11 (2009) 6592-6599. DOI:10.1039/b905841a |
| [8] |
(a) J.W. Peters, W.N. Lanzilotta, B.J. Lemon, L.C. Seefeldt, Science 282 (1998) 1853-1858; (b) Y. Nicolet, C. Piras, J.C. Fontecilla-Camps, et al., Structure 7 (1999) 13-23. |
| [9] |
(a) C. Tard, C.J. Pickett, Chem. Rev. 109 (2009) 2245-2274; (b) L.Z. Wu, B. Chen, Z.J. Li, C.H. Tung, Acc. Chem. Res. 47 (2014) 2177-2185; (c) S.J. Borg, T. Behrsing, C.J. Pickett, et al., J. Am. Chem. Soc. 126 (2004) 16988-16999; (d) F. Wang, W.G. Wang, L.Z. Wu, et al., ACS Catal. 2 (2012) 407-416; (e) Y. Na, M. Wang, L. Sun, et al., Inorg. Chem. 47 (2008) 2805-2810; (f) L.J. Antila, P. Ghamgosar, L. Hammarström, et al., ACS Energy Lett. 1 (2016) 1106-1111; (g) A.M. Brown, L.J. Antila, L. Hammarström, et al., J. Am. Chem. Soc.138 (2016) 8060-8063. |
| [10] |
(a) L.C. Song, Z.Y. Yang, Q.M. Hu, et al., Organometallics 24 (2005) 6126-6135; (b) L.C. Song, Q.L. Li, H.B. Song, et al., Dalton Trans. 42 (2013) 1612-1626; (c) W.G. Wang, H.Y. Wang, L.Z. Wu, et al., Dalton Trans. 15 (2009) 2712-2720; (d) P. Li, M. Wang, L. Sun, et al., Eur. J. Inorg. Chem. 2007 (2007) 3718-3727; (e) C.M. Thomas, O. Rüdiger, M.Y. Darensbourg, et al., Organometallics 26 (2007) 3976-3984; (f) S. Ezzaher, J.F. Capon, J. Talarmin, Inorg. Chem. 8 (2009) 2-4; (g) L.C. Song, F.X. Luo, B.B. Liu, et al., Organometallics 35 (2016) 1399-1408; (h) M. Cheng, M. Wang, D. Zheng, L. Sun, Dalton Trans. 45 (2016) 17687-17696; (i) D. Zheng, N. Wang, L. Sun, et al., J. Am. Chem. Soc. 136 (2014) 16817-16823. |
| [11] |
(a) M. Razavet, X. Liu, C.J. Pickett, Dalton Trans. 236 (2003) 586-595; (b) U.P. Apfel, Y. Halpin, H. Görls, J.G. Vos, W. Weigand, Eur. J. Inorg. Chem. 2011 (2011) 581-588. |
| [12] |
(a) J.W. Tye, J. Lee, M.Y. Darensbourg, Inorg. Chem. 44 (2005) 5550-5552; (b) D. Morvan, J.F. Capon, F. Gloaguen, et al., Organometallics 26 (2017) 2042-2052; (c) J.F. Capon, S.E. Hassnaoui, F. Gloaguen, et al., Organometallics 24 (2005) 2020-2022. |
| [13] |
(a) X. Zhao, I.P. Georgakaki, M.Y. Darensbourg, et al., J. Am. Chem. Soc. 123 (2001) 9710-9711; (b) J.A. Wright, C.J. Pickett, Chem. Commun. 45 (2009) 5719-5721; (c) A. Jablonskyte, J.A. Wright, C.J. Pickett, Dalton Trans. 39 (2010) 3026-3034. |
| [14] |
(a) W. Wang, M.J. Nilges, T.B. Rauchfuss, M. Stein, J. Am. Chem. Soc.135 (2013) 3633-3639; (b) A. Jablonskyte, J.A. Wright, C.J. Pickett, et al., J. Am. Chem. Soc. 133 (2011) 18606. |
| [15] |
(a) S. Ezzaher, J.F. Capon, N. Kervarec, Inorg. Chem. 46 (2007) 3426-3428; (b) B.E. Barton, T.B. Rauchfuss, S.R. Wilson, et al., DaltonTrans. 39 (2010) 3011-3019. |
| [16] |
(a) R. Zaffaroni, T.B. Rauchfuss, D.L. Gray, et al., J. Am. Chem. Soc. 134 (2012) 19260-19269; (b) J.I.V.D. Vlugt, T.B. Rauchfuss, C.M. Whaley, S.R. Wilson, J. Am. Chem. Soc.127 (2005) 16012-16013. |
| [17] |
X. Yu, C.H. Tung, T.B. Rauchfuss, et al., Organometallics 36 (2017) 2245-2253. DOI:10.1021/acs.organomet.7b00297 |
| [18] |
(a) S. Munery, J.F. Capon, L. DeGioia, et al., Chem.-Eur. J. 19 (2013) 15458-15461; (b) W. Wang, T.B. Rauchfuss, C.E. Moore, et al., Chem.-Eur. J. 19 (2013) 15476-15479; (c) R. Goy, L. Bertini, C. Elleouet, et al., Dalton Trans. 44 (2015) 1690-1699. |
| [19] |
(a) T. Liu, M.Y. Darensbourg, J. Am. Chem. Soc. 129 (2007) 7008-7009; (b) C.H. Hsieh, O.F. Erdem, S.D. Harman, et al., J. Am. Chem. Soc. 134 (2012) 13089-13102. |
| [20] |
A.K. Justice, T.B. Rauchfuss, S.R. Wilson, Angew. Chem. Int. Ed. 46 (2007) 6152-6154. |
| [21] |
(a) J.M. Camara, T.B. Rauchfuss, Nat. Chem. 4 (2011) 26-30; (b) N. Wang, M. Wang, L. Sun, et al., J. Am. Chem. Soc.135 (2013) 13688-13691; (c) D. Zheng, M. Wang, L. Sun, et al., Chem. Commun. 50 (2014) 9255-9258. |
| [22] |
(a) D. Chouffai, G. Zampella, J.F. Capon, et al., Organometallics 31 (2012) 1082-1091; (b) D. Chouffai, G. Zampella, J.F. Capon, et al., Inorg. Chem. 50 (2011) 12575-12585; (c) C.M. Thomas, T. Liu, M.B. Hall, M.Y. Darensbourg, Chem. Commun.13 (2008) 1563; (d) A.K. Justice, M.J. Nilges, T.B. Rauchfuss, et al., J. Am. Chem. Soc. 130 (2008) 5293-5301; (e) A.K. Justice, L.D. Gioia, T.B. Rauchfuss, et al., Inorg. Chem. 47 (2008) 7405-7414. |
| [23] |
(a) A. Jablonskyte, J.A. Wright, C.J. Pickett, et al., Angew. Chem. Int. Ed. 53 (2014) 10143-10146; (b) M. Razavet, S.J. Borg, C.J. Pickett, et al., Chem. Commun. 7 (2002) 700-701; (c) C. Tard, X. Liu, C.J. Pickett, et al., Nature 433 (2005) 610-613; (d) M. Razavet, S.C. Davies, D.L. Hughes, C.J. Pickett, Chem. Commun. 9 (2001) 847-848; (e) J. Liu, A. Zhang, H. Song, et al., Chin. Chem. Lett. 29 (2018) 949-953. |
| [24] |
A.K. Justice, G. Zampella, T.B. Rauchfuss, et al., Inorg. Chem. 46 (2007) 1655-1664. |
| [25] |
F.I. Adam, G. Hogarth, I. Richards, B.E. Sanchez, Dalton Trans. 24 (2007) 2495-2498. |
| [26] |
M.L. Singleton, N. Bhuvanesh, J.H. Reibenspies, M.Y. Darensbourg, Angew. Chem. 47 (2008) 9492-9495. DOI:10.1002/anie.v47:49 |
| [27] |
W. Wang, T.B. Rauchfuss, L. Bertini, G. Zampella, J. Am. Chem. Soc. 134 (2012) 4525-4528. DOI:10.1021/ja211778j |
| [28] |
N.G. Connelly, W.E. Geiger, Chem. Rev. 96 (1996) 877-910. DOI:10.1021/cr940053x |
| [29] |
D. Schilter, S. Hammes-Schiffer, T.B. Rauchfuss, et al., Chem. Rev. 116 (2016) 8693-8749. |
| [30] |
S. Lounissi, G. Zampella, J.F. Capon, et al., Chem.-Eur. J. 18 (2012) 11123-11138. DOI:10.1002/chem.201201087 |
| [31] |
T. Fujinaga, I. Sakamoto, J. Electroanal. Chem. 85 (1977) 185-201. DOI:10.1016/S0022-0728(77)80163-0 |
| [32] |
K. Abdur-Rashid, T.P. Fong, R.H. Morris, et al., J. Am. Chem. Soc. 122 (2000) 9155-9171. |
| [33] |
G.A.N. Felton, R.S. Glass, D.L. Lichtenberger, D.H. Evans, Inorg. Chem. 45 (2006) 9181-9184. DOI:10.1021/ic060984e |
| [34] |
I. Kaljurand, A. Kütt, L. Sooväli, et al., J. Org. Chem. 70 (2005) 1019-1028. |
| [35] |
M.E. Carroll, B.E. Barton, T.B. Rauchfuss, P.J. Carroll, J. Am. Chem. Soc. 134 (2012) 18843-18852. DOI:10.1021/ja309216v |
| [36] |
R.H. Morris, Chem. Rev. 116 (2016) 8588-8654. DOI:10.1021/acs.chemrev.5b00695 |
| [37] |
E. Block, G. Oforiokai, J. Zubieta, J. Am. Chem. Soc. 111 (1989) 2327-2329. DOI:10.1021/ja00188a071 |