 2018, Vol. 29
2018, Vol. 29 



As one of the most prestigious organocatalysts, cinchona alkaloid has shown remarkable catalytic activities in a wide range of transformations, promoting carbon-carbon and carbon-heteroatom bond formations [1]. Particularly, the cinchona alkaloid catalysts have served as one of the major work horses in the asymmetric Michael addition reactions, achieving the desired reactivity and enantioselectivity [2]. This reaction provides an efficient strategy for the construction of small molecules, and has received considerable interests in the synthetic community.
The cinchona alkaloid derivatives contain a natural chiral pocket with multiple hydrogen-bonding sites (Fig. 1). These structural features make the cinchona alkaloid one of the key privileged organocatalysts since it can effectively bind the substrate in a close proximity. The hydrogen-bonding network provides a specific chiral environment for the substrate, which leads to the enantioselective transformations. Although the underlying working mechanism for cinchona alkaloid is clear, the molecular level of understandings of the chiral hydrogenbonding networks, and especially the origins of the observed enantioselectivities, are still primitive.

|
Download:
|
| Fig. 1. General structure of cinchona alkaloid catalysts and potential hydrogenbonding sites (highlighted in blue). | |
{kind=link}
The lack of predictive model for the cinchona alkaloid derivatives presents a significant challenge for future rational designs of related transformations. To elucidate the molecular level mechanism and the controlling factors of reactivities and enantioselectivities, remarkable efforts have been dedicated by the computational chemists to study the cinchona alkaloidcatalyzed asymmetric Michael additions. Categorized by the types of bond formations, this review summarizes the fruitful advances of this area.
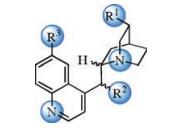
2. Computational studies of cinchona alkaloid-catalyzed Michael addition for C-C bond formationBased on their own experimental studies [3], Wang and coworkers studied the mechanism of the cinchona thioureacatalyzed asymmetric Michael addition of α, β-unsaturated γ-butyrolactam with chalcone (Fig. 2), using NMR and density functional theory (DFT) calculations [4]. Using the experimental catalyst 1 as the model for calculations, they discovered that the C-C bond formation step is the enantioselectivity-determining step, with TS1 leading to the favorable enantiomer. In TS1, the catalyst uses both the thiourea N-H and the quinuclidinium to interact with the nucleophile lactam, and simultaneously activates the electrophile chalcone with the other thiourea N-H (Fig. 2). The competing transition state for the minor enantiomer TS2 is 3.9 kcal/mol less favorable as compared to TS1. The transition states that lead to the diastereomers (TS3 and TS4) were also located, and are higher in free energies comparing with TS1 (Fig. 2). These computational results agree well with the experimentally observed selectivities [3]. The enantioselectivity is rationalized by both the electronic and steric effects, TS1 has strong hydrogenbonding interactions between the catalyst and chalcone which stabilizes this favorable transition state, while TS2 has the acyl moiety of the chalcone proximal to the quinuclidinium moiety of the catalyst which leads to significant steric repulsions.

|
Download:
|
| Fig. 2. Experimental [3] and computational [4] studies of cinchona thiourea-catalyzed asymmetric Michael addition of α, β-unsaturated γ-butyrolactam with chalcone. The hydrogens are omitted in the transition state diagrams except the ones involved in the hydrogen-bonding interactions. | |
{kind=link}
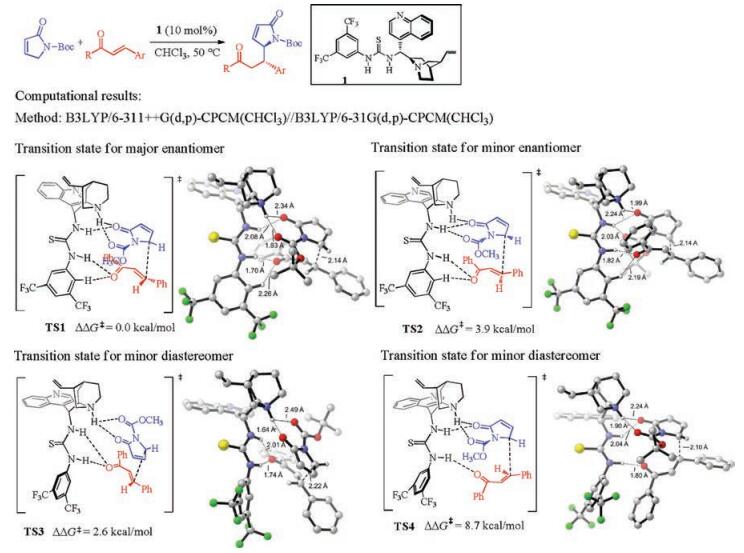
Focusing on the experimental results of cinchona thioureacatalyzed Michael addition of nitroalkanes to enones [5], Grayson thoroughly studied the reaction mechanism and origins of enantioselectivity with DFT calculations (Fig. 3) [6]. It is discovered that the favored transition states have both the catalyst thiourea N-Hs of the catalyst 2 interacting with the deprotonated nitromethane, and the quinuclidinium has a hydrogen-bonding interaction with the enone (TS5 and TS6, Fig. 3). TS5 is 5.4 kcal/mol more stable than the competing TS6, which is consistent with the experimental enantioselectivities [5]. TS6 is unfavorable due to the s-trans geometry of the enone substrate, and the lack of the favorable π–π stacking between the phenyl group of the enone substrate and the catalyst CF3-substituted aryl group that is presented in TS5.

|
Download:
|
| Fig. 3. Experimental [5] and computational [6] studies of cinchona thiourea-catalyzed asymmetric Michael addition of nitroalkanes with enones. The hydrogens are omitted in the transition state diagrams except the ones involved in hydrogen-bonding interactions. | |
{kind=link}
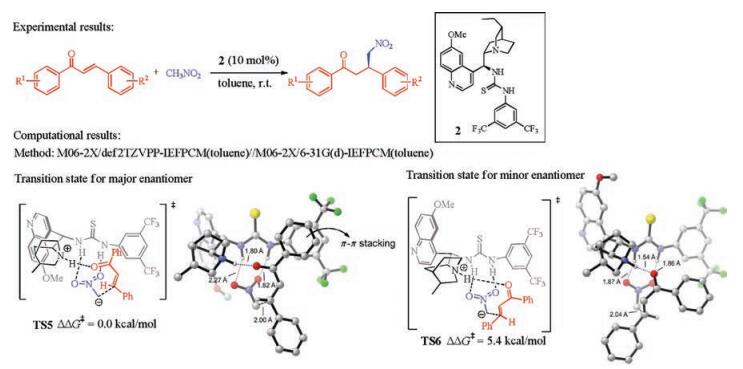
Dang and co-workers studied the reaction mechanism of the cinchona thiourea-amine catalyzed Michael addition of malonate with α, β-unsaturated ketones, using pentane-2, 4-dione and acrylaldehyde as the substrate in the computations (Fig. 4) [7, 8]. In this work, an interesting "triple activation" model is discovered (TS7, Fig. 4). The cinchona thiourea-amine catalyst has the protonated primary amine moiety interacting with the deprotonated pentane-2, 4-dione substrate, and the thiourea N-Hs forming hydrogen bonds with the acrylaldehyde. In addition, the quinuclidine group is responsible for the enantioselectivity. Therefore, all the three moieties of the organocatalyst are essential for the reactivity and enantioselectivity.

|
Download:
|
| Fig. 4. Experimental [7] and computational [8] studies of cinchona thiourea-amine catalyzed asymmetric Michael addition of malonate with α, β-unsaturated ketones. The hydrogens are omitted in the transition state diagram except the ones involved in hydrogen-bonding interactions. | |
{kind=link}
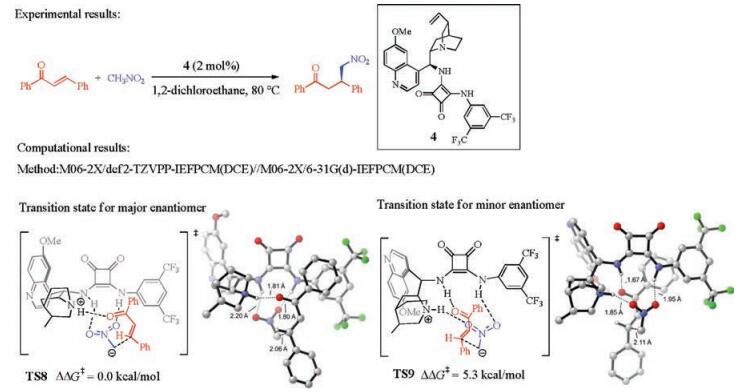
In addition to the cinchona thiourea catalysts, other types of cinchona catalysts have also been explored computationally for the asymmetric Michael addition involving C-C bond formations. Grayson studied the cinchona squaramide-catalyzed asymmetric Michael addition of nitroalkanes to enones (Fig. 5) [6]. The C-C bond formation is again the enantioselectivity-determining step, and the favorable transition state TS8 has two types of hydrogenbonding interactions. One exists between the squaramide and the deprotonated nitromethane, and the other is between the quinuclidinium and the enone carbonyl group. This is consistent with the above mechanistic model of cinchona thiourea for the same reaction (TS5, Fig. 3). The transition state leading to the unfavorable enantiomer is 5.3 kcal/mol less favorable (TS8 vs. TS9, Fig. 5), which is consistent with the corresponding results with thiourea (Fig. 3).

|
Download:
|
| Fig. 5. Experimental [9] and computational [6] studies of cinchona squaramide-catalyzed asymmetric Michael addition of nitroalkanes with enones. The hydrogens are omitted in the transition state diagram except the ones involved in hydrogen-bonding interactions. | |
{kind=link}
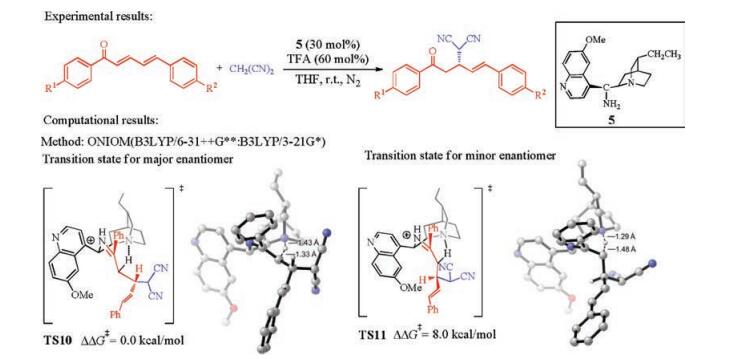
Kim and co-workers investigated the mechanism and origins of enantioselectivities of cinchona amine-catalyzed asymmetric 1, 4-Miachel addition of malononitrile to 1, 5-diphenylpenta-2, 4-dien-1-one (Fig. 6) [10]. In contrast to the previous studies, they discovered that the proton transfer step is the enantioselectivity-determining step (Fig. 6). This step occurs after the C-C bond formation, and the proton transfers from the quinuclidinium to the α-position of ketone. In the favored transition state TS10, the bulky δ-phenyl moiety with the CH(CN)2 substituent is distal from the quinuclidine group of the catalyst, while the same moiety is proximal to the quinuclidine group of the catalyst in the competing TS11 (Fig. 6). This leads to the 8.0 kcal/mol Gibbs free energy difference between the two transition states, and the computational results are consistent with the experimental selectivity [11].

|
Download:
|
| Fig. 6. Experimental [11] and computational [10] studies of cinchona amine-catalyzed asymmetric Michael addition of malononitrile to 1, 5-diphenylpenta-2, 4-dien-1-one. The hydrogens are omitted in the transition state diagram except the transferring proton. | |
{kind=link}
3. Computational studies of cinchona alkaloid-catalyzed Michael addition for C-S bond formation
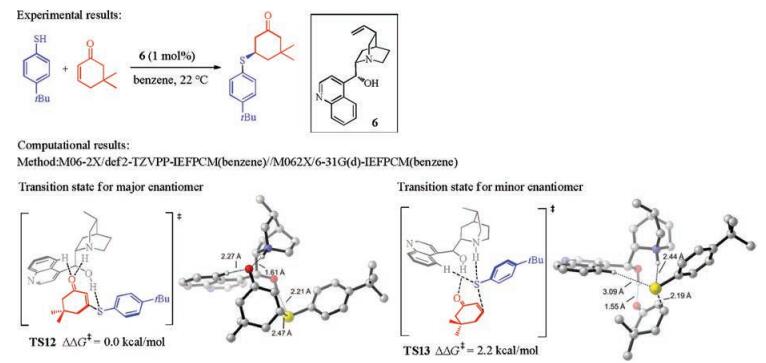
Aside from the carbon nucleophiles, the asymmetric Michael additions with sulfur nucleophiles have also been thoroughly studied in a number of computational investigations. Houk and coworkers studied the cinchona alkaloid-catalyzed conjugate additions of aromatic thiols to cycloalkenones, and revealed the activation mode and the origins of enantioselectivities (Fig. 7) [12]. The favorable activation mode (TS12, Fig. 7) includes the hydrogenbonding interaction between the hydroxyl group and the attacking sulfur atom, and the carbonyl of ketone interacts with the quinuclidinium moiety. In the transition state leading to the minor enantiomer (TS13, Fig. 7), the partners of hydrogen-bonding interactions are switched, and the sulfur is interacting with the quinuclidinium. The calculated enantioselectivity is 2.2 kcal/mol, which agrees well with the experimental results [13], and this enantioselectivity is rationalized based on the strength of hydrogen-bonding interactions between the catalyst and the substrates.

|
Download:
|
| Fig. 7. Experimental [13] and computational [12] studies of cinchona alkaloid-catalyzed conjugate additions of aromatic thiols to cycloalkenones. The hydrogens are omitted in the transition state diagram except the ones involved in hydrogen-bonding interactions. | |
{kind=link}
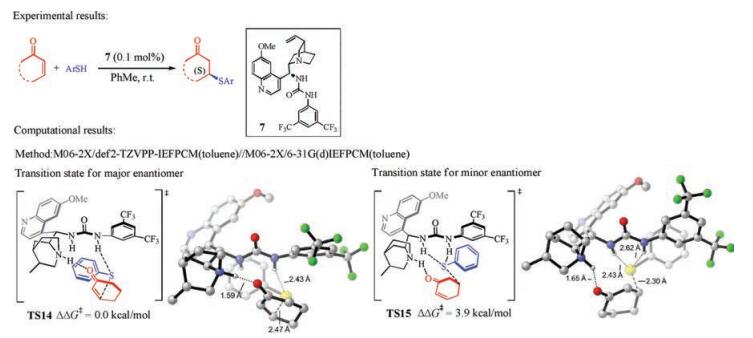
Houk and Grayson used DFT calculations to study the mechanism and origins of enantioselectivities of the cinchona urea-catalyzed asymmetric Michael addition of aryl thiols to cycloalkenones (Fig. 8) [14, 15]. A Brønsted acid-hydrogen bonding model is discovered. The C-S bond formation determines the enantioselectivity. In the favored transition state TS14, the cinchona urea uses the urea N-H to interact with the aryl thiolate, and the cycloalkenone has a strong hydrogen-bonding interaction with the quinuclidinium moiety (Fig. 8). Comparing the two competing transition states, a 3.9 kcal/mol preference is found, which is consistent with the experimental selectivity. This enantioselectivity is rationalized by two effects. First, TS15 has a weaker interaction between the quinuclidinium cation and the enone oxygen as compared to TS14. In addition, the favored transition state TS14 has a staggered conformation of the forming C-S bond, while this conformation of the same forming bond in TS15 adopts an eclipsed conformation.

|
Download:
|
| Fig. 8. Experimental [14] and computational [15] studies of cinchona urea-catalyzed asymmetric Michael addition of aryl thiols to cycloalkenones. The hydrogens are omitted in the transition state diagram except the ones involved in hydrogen-bonding interactions. | |
{kind=link}
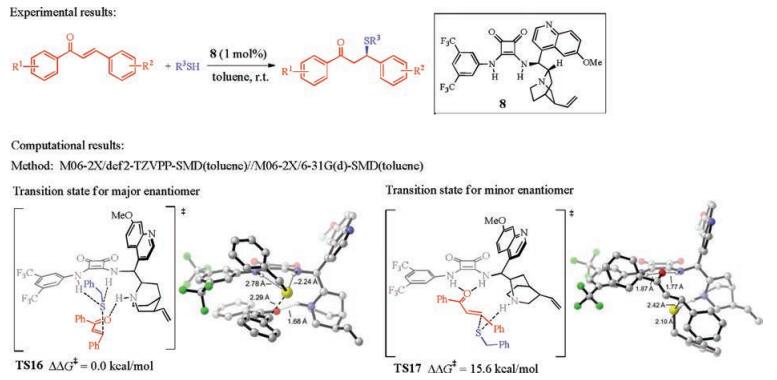
Recently, Wong and co-workers studied the activation model and the origins of enantioselectivity of the cinchona squaramidecatalyzed asymmetric Michael addition of benzyl thiols to tran-schalcone (Fig. 9) [16, 17]. The DFT calculations revealed a similar activation model as the one that Houk and co-workers has discovered (Fig. 8) [15]. In the enantioselectivity-determining C-S bond formation step, the favored transition state TS16 has the squaramide N-Hs interacting with the benzyl thiolate, and the quinuclidinium moiety has an additional hydrogen bond with the chalcone oxygen (Fig. 9). The distortion/interaction analysis elucidated the origins of the enantioselectivity. The favorable interactions between the catalyst and substrates in the favored TS16 are the major reasons that stabilize this transition state and differentiate the two enantiomers. The noncovalent interaction analysis provided further support for this rationalization.

|
Download:
|
| Fig. 9. Experimental [16] and computational [17] studies of cinchona squaramide-catalyzed asymmetric Michael addition of benzyl thiols to trans-chalcone. The hydrogens are omitted in the transition state diagram except the ones involved in hydrogen-bonding interactions. | |
{kind=link}
4. Conclusion and outlook
This review summarizes the fruitful computational studies on the mechanism and enantioselectivities of cinchona alkaloidcatalyzed asymmetric Michael additions. These remarkable works elucidated the key activation models of a wide range of substrates and catalysts, provided the fundamental understandings for cinchona alkaloid catalysts. The cinchona alkaloid catalysts generally work as a bifunctional catalyst, which activates the nucleophile and electrophile simultaneously. Such catalyst-substrate interactions are mostly hydrogen-bonding interactions, but additional noncovalent interactions can also play important roles. Through the multiple hydrogen-bonding interactions, the substrates adopt the chiral environment of the cinchona alkaloid, which eventually leads to the desired enantioselectivity.
The discoveries included in this review provide a general perspective of this exciting area, but a number of important directions still require future computational studies for the cinchona alkaloid catalysts. Most of the current computational studies focused on the rationalization of experimentally observed reactivities and selectivities. The combined computational and experimental studies can allow a unique opportunity for the rational design of cinchona alkaloid catalysts with target catalytic properties. In addition, the current calculations mainly rely on the transition state theory and the static picture of the reaction process, while the dynamics of the hydrogen-bonding network is neglected in most cases [18]. The dynamic behaviors of these hydrogen-bonding networks can potentially affect the enantioselectivity, and require further elaborations. We hope and believe that more impressive investigations in the area of cinchona alkaloid catalysts will emerge, allowing the molecular level of design for future organocatalysts.
AcknowledgmentsThis paper is dedicated to Professor Kendall N. Houk on the occasion of his 75th birthday. Financial support from the National Natural Science Foundation of China (NSFC, No. 21702182), the Chinese "Thousand Youth Talents Plan", "Fundamental Research Funds for the Central Universities", and Zhejiang University is gratefully acknowledged.
| [1] |
(a) Y. Takemoto, Chem. Pharm. Bull. 58 (2010) 593-601; (b) P. Melchiorre, Angew. Chem. Int. Ed. 51 (2012) 9748-9770; (c) Y.H. Lam, M.N. Grayson, M.C. Holland, A. Simon, K.N. Houk, Acc. Chem. Res. 49 (2016) 750-762; (d) G. Tanriver, B. Dedeoglu, S. Catak, V. Aviyente, Acc. Chem. Res. 49 (2016) 1250-1262. |
| [2] |
(a) C. Palomo, M. Oiarbide, R. López, Chem. Soc. Rev. 38 (2009) 632-653; (b) P. Chauhan, S. Mahajan, U. Kaya, D. Hack, D. Enders, Adv. Synth. Catal. 357 (2015) 253-281; (c) K. Kacprzak, J. Gawronski, Synthesis 7 (2001) 961-998; (d) A.E. Nibbs, K.A. Scheidt, Eur. J. Org. Chem. 2012 (2012) 449-462; (e) X. Han, H.B. Zhou, C. Dong, Chem. Rec. 16 (2016) 897-906; (f) B.L. Zhao, J.H. Li, D.M. Du, Chem. Rec. 17 (2017) 902-1069. |
| [3] |
Y. Zhang, Y.L. Shao, H.S. Xu, W. Wang, J. Org. Chem. 76 (2011) 1472-1474. DOI:10.1021/jo102223v |
| [4] |
J.L. Zhu, Y. Zhang, C. Liu, A.M. Zheng, W. Wang, J. Org. Chem. 77 (2012) 9813-9825. DOI:10.1021/jo302133n |
| [5] |
B. Vakulya, S. Varga, A. Csámpai, T. Soós, Org. Lett. 7 (2005) 1067-1969. DOI:10.1021/ol050019c |
| [6] |
M.N. Grayson, J. Org. Chem. 82 (2017) 4396-4401. DOI:10.1021/acs.joc.7b00521 |
| [7] |
P. Li, S. Wen, F. Yu, et al., Org. Lett. 11 (2009) 753-756. DOI:10.1021/ol802892h |
| [8] |
M.J. Zhou, Y. Li, L. Dang, Asian J. Org. Chem. 4 (2015) 904-911. DOI:10.1002/ajoc.v4.9 |
| [9] |
W. Yang, D.M. Du, Org. Lett. 12 (2010) 5450-5453. DOI:10.1021/ol102294g |
| [10] |
Z.S. Su, H.W. Lee, C.K. Kim, Eur. J. Org. Chem. 2013 (2013) 1706-1715. |
| [11] |
C.G. Oliva, A.M.S. Silva, D.I.S.P. Resende, F.A.A. Paz, J.A.S. Cavaleiro, Eur. J. Org. Chem. 2010 (2010) 3449-3458. |
| [12] |
M.N. Grayson, K.N. Houk, J. Am. Chem. Soc. 138 (2016) 1170-1173. DOI:10.1021/jacs.5b13275 |
| [13] |
H. Hiemstra, H. Wynberg, J. Am. Chem. Soc. 103 (1981) 417-430. DOI:10.1021/ja00392a029 |
| [14] |
N.K. Rana, S. Selvakumar, V.K. Singh, J. Org. Chem. 75 (2010) 2089-2091. DOI:10.1021/jo902634a |
| [15] |
M.N. Grayson, K.N. Houk, J. Am. Chem. Soc. 138 (2016) 9041-9044. DOI:10.1021/jacs.6b05074 |
| [16] |
L. Dai, S.X. Wang, F.E. Chen, Adv. Synth. Catal. 352 (2010) 2137-2141. DOI:10.1002/adsc.201000334 |
| [17] |
J.L. Guo, M.W. Wong, J. Org. Chem. 82 (2017) 4362-4368. DOI:10.1021/acs.joc.7b00388 |
| [18] |
M.N. Grayson, Z. Yang, K.N. Houk, J. Am. Chem. Soc. 139 (2017) 7717-7720. DOI:10.1021/jacs.7b03847 |