 2018, Vol. 29
2018, Vol. 29 

 ,
Jian Yina
,
Jian Yinab Wuxi School of Medicine, Jiangnan University, Wuxi 214122, China
Carbohydrates not only serve as energy sources and stand structural elements, but also play a very important role as signalling molecules in cells. Their biological significance includes cell proliferation, differentiation, migration, cell-cell adhesion, trafficking, and receptor binding and activation [1, 2]. Recently, intensive attention has been drawn to the synthesis of oligosaccharides, the main component of capsular polysaccharides (CPS) and lipopolysaccharides (LPS) on the bacterial cells related to immune responses [3-7]. For example, chemically synthesized polysaccharides have been employed in the development of carbohydrate-based vaccines [8-11].
Epimerization of secondary alcohols on carbohydrates towards the corresponding converted structuresare ubiquitous in carbohydrate chemistry. For example, the synthesis of amino sugars, thio-sugars, halogen-sugars and rare sugars can be obtained by inversion methods from common saccharides. The "controlled inversion strategies" in carbohydrate synthesis [12], playing a key role in carbohydrate-related pharmaceutical research and development, are summarized in this review, including mechanisms and applications. Several different inversion strategies have been developed based on nucleophilic reactions of sulfonates, sequential oxidation/reduction routes, the Mitsunobu reaction with triphenylphosphine (PPh3), enzymatic inversion methods and epimerization by non-classical acetalization.
2. Inversion via sulfonyl groupsThe utilization of the sulfonyl groups for inverting a given hydroxyl group is a well-known method in carbohydrate chemistry, followed by SN2 displacement with a variety of reagents, such as acetate, benzoate, thioacetate, a nucleophilic acetamido group, and others. The direct replacement of sulfonyl groups by nucleophilic reagents undergoes an SN2 mechanism, which can be traced to the Richardson-Hough rules recently updated by Hale et al. [13, 14]. Among these epimerization reactions, the nitritemediated Lattrell-Dax method discovered by Lattrell and Lohaus in 1973 is an efficient method to generate inverted configurations [15, 16]. Converted configurations with unprotected hydroxyl groups can be used to prepare various derivatives by introducing different protecting groups. Recently, Dong discussed stereo specific ester activation in nitrite-mediated carbohydrate epimerization [17]. It was suggested that good yields can be obtained from these nitrite-mediated inversions only if the neighbouring equatorial esters are utilized as protecting groups. When the ester protecting groups were replaced by benzyl (Bn)/benzylidene groups, a mixture of different products was produced instead. Due to the mild reaction conditions and readily good yields obtained in most cases, the Lattrell-Dax epimerization has been widely used in carbohydrate chemistry in recent years.
Nucleophilic displacement reactions via sulfonyl groups have been applied for the synthesis of various types of sugars, such as amino sugars, thio-sugars and rare sugars obtained from monosaccharide which were appropriately protected by leaving groups, such as mesylate (OMs), tosylate (OTs), and triflate. Compared with triflates, which are formed by the reaction of an alcohol with triflic anhydride (Tf2O) in pyridine (Py), mesylates and tosylates are economical to obtain by the treatment of an alcohol with sulfonyl chloride in Py. Moreover, unlike mesylates and tosylates, triflates are inappropriate to be purified by silica gel column chromatography; however, they can be up to 103 times more reactive towards substituents than tosylates. Two factors contribute to the difficulty involved in SN2 reactions with sulfonyl groups on carbohydrate substrates. One is the steric hindrance of the heavily oxygenated carbohydrate carbon ring. The other is the participation of nearby hydroxyls, protecting groups and ether oxygens on the reactive carbohydrate, leading to rearranged or double-inverted products [18-20].
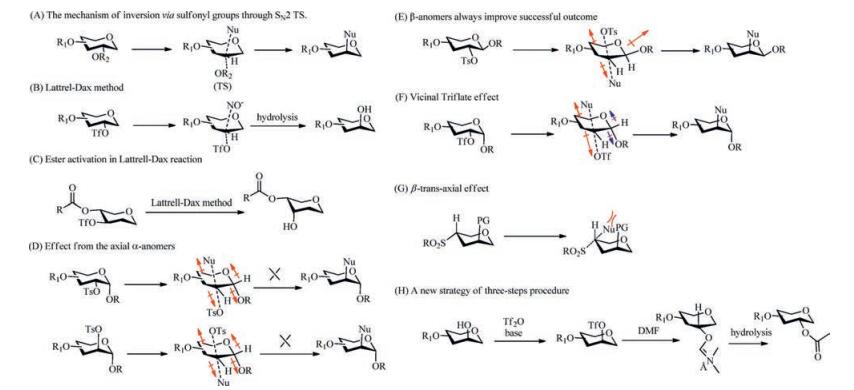
2.1. Mechanism of inversion via sulfonyl groupsNucleophilic substitution reactions via sulfonyl groups normally proceed via an SN2 mechanism, resulting in the inversion of configuration. As shown in Scheme 1, the crucial step in this progress is the establishment of the SN2 transition state (TS)(Scheme 1A), which contains two highly polar bonds related to the process of formation and degeneration [20]. The success of the reaction performing smoothly via sulfonyl groups is determined by two factors: polarity and steric hindrance [14]. The polarity of the SN2 reaction is influenced by the nucleophilicity of the incoming nucleophile, the nature of the leaving group, the polarity of the solvent and the steric electronic effect.

|
Download:
|
| Scheme 1. Mechanisms of inversion via sulfonyl groups. (A) The mechanism of inversion via sulfonyl groups through SN2 TS. (B) Lattrel-Dax method. (C) Ester activation in Lattrell-Dax reaction. (D) Effect from the axial α-anomers. (E) β-anomers always improve successful outcome. (F) Vicinal Triflate effect. (G) β-trans-axial effect. (H) A new strategy of three-step procedure. | |
{kind=link}
Among the various types of epimerization reactions, the nitritemediated Lattrell-Dax method is consistent with the SN2 displacement via sulfonyl groups introduced above. In this epimerization reaction, the anion is used as a nucleophile to form an unstable intermediate, which then undergoes the hydrolysis of nitrite bond, resulting in the epi-hydroxyl structure (Scheme 1B)[15]. The neighbouring equatorial ester group has been reported to be essential for a good yield in the Lattrell-Dax epimerization by Dong et al. (Scheme 1C) [17, 21-23]. Due to its efficiency and convenience, this strategy has beenwidely used in carbohydrate synthesis.
2.2. The rules of inversion via sulfonyl groupsFor most SN2 reactions, steric hindrance can critically hamper the reaction due to the considerable increase of the activation energy for the SN2 process [20]. Therefore, several rules are generalized according to experimental data (The red arrow indicates the polar bond, and the permanent dipole may impede the development of TS). The first rule is that α-D-pyranoside C(2)-OTs and —OMs have difficulty undergoing SN2 displacement because of the effect from the axial α-glycoside at C(1)(Scheme 1D). Successful outcomes normally can be obtained with β-anomers (Scheme 1E) [13]. The second rule is that a C(2)-O-triflate glycoside could be a better choice than the C(2)-OTs or C(2)-OMs species because of the Vicinal Triflate Effect termed by Hale et al. [13]. The Vicinal Triflate Effect has a diminishing effect on the magnitude of the adjacent repulsive dipole between the reacting carbon and vicinal protecting groups by the electron-withdrawing O-triflate group in the developing SN2 TS (Scheme 1F) [24].
The last rule is that pyranoside 3- and 4-sulfonates can usually undergo successful reactions, except under two conditions: (1) When there is a vicinal axial electronegative substituent next to the sulfonate group, (2) When there is a β-trans-axial substituent relative to the C—OSO2 R group (Scheme 1G).
Noteworthily, Hosoya et al. recently reported a new method for the stereo inversion of secondary alcohols on carbocyclic derivatives, including the triflation of alcohols, nucleophilic displacement by treatment with aqueous N, N-dimethylformamide (DMF) and methanolysis [25]. This three-step procedure is unique for the oxygenation displacement proceeding under acidic conditions, providing a new option for the stereo inversion of the hydroxyl group. Occasionally, SN2 displacements by sulfonyl groups cause competing reactions resulting in elimination and secondary flipping products [18, 26]. These side reactions were proposed to be avoided by the strong superior leaving group ability of the triflate and low Brønsted basicity of DMF, which is used as a nucleophile to form a configuration inversion (Scheme 1H) [27, 28].
2.3. Applications of inversion via sulfonyl groupsIn recent decades, applications of inversion via sulfonyl groups followed by substitution reactions in carbohydrate chemistry have been divided into four types according to the substituents and the effects depicted in Scheme 2A: The preparation of amino sugars, the preparation of rare sugars, the synthesis of β-D-mannosides and talosides and the preparation of halogen sugars, thio-sugars, seleno-sugars, etc.

|
Download:
|
| Scheme 2. Applications of inversion strategy via sulfonyl groups. (A) Different applications of inversions via sulfonyl groups. (B) Synthsis of amino-sugar 2, 4 by inversion strategy via sulfonyl groups. (C) Synthesis of amino-sugar containing β-glycoside 6, 8 by inversion strategy via sulfonyl groups. | |
{kind=link}
Amino sugars are of vital importance because they contribute to living organisms and serve as components of glycoproteins, glycolipids, and proteoglycans. A frequently used approach to introduce amino groups is the application of SN2 displacement reactions, in which an appropriate leaving group (i.e., sulfonate or halide) is replaced by a nitrogen-containing nucleophile, such as ammonia, hydrazine, or phthalimide (Phth) or azide ions. To date, azide ions have been the most commonly used nucleophilic reagents in substitution reactions for the preparation of amino sugars. 2-Amino-2-deoxyhexoses are one of the most important sugars found in nature [29]. 2-Azide-2-deoxy-D-mannose can be obtained by conversion of glucose that is abundantly available [30, 31].
Recently, Seeberger et al. developed a de novo synthesis of the bacterial building block D-Bacillosamine (Bac) 2 (Scheme 2B), which is important for bacterial adherence and invasion expressed on bacteria, such as the cell surface of N. gonorrhoea, inverted from the prepared D-fucosamine derivative 1 (Scheme 2B) with 47% yield over two steps [32-34]. Similarly, to introduce an azide group, 3-azido-galactoside was synthesized by double inversions at C3 from methyl-2-acetyl (Ac)-4, 6-O-benzylidene-β-D-galactopyranoside in a yield of 40% over three steps [35]. The synthesis of D-glycero-D-gulo sialic acid 4 (Scheme 2B) is a good example of the synthesis of an azide derivative, inverted from 5-O-triflate 3 in 74% yield (Scheme 2B) [36]. Very recently, Kulkarni et al. developed a general and expedient methodology to obtain a variety of deoxy amino L-sugars from L-rhamnose and L-fucose [37]. Based on orthogonally protected deoxy amino L-sugar building blocks, the first total syntheses of the amino-linker-attached, conjugationready oligosaccharides of O-PS, Yersinia enterocolitica O:50 strain 3229 and Pseudomonas chlororaphis subsp. aureofaciens strain M71, were developed. Selective inversions at desired positions are also allowed by the regioselective monotriflations at O2, O3 and O4 of L-fucose/L-rhamnose.
The epimerization of 2-C-β-glucosides is a well-known method to mitigate the difficultly of β-mannoside construction in carbohydrate synthesis [38]. This inversion strategy invented by Miljkovic et al. has been widely applied due to the easily controlled acceptor in glycosylation and the satisfactory outcome [39, 40]. The 2-mesyloxy group was displaced with a benzoate, providing bmannoside in 62% yield. 1, 2-Trans-β-glucosides can be obtained by the glycosylation of C-2 acetyl-glucose donors because of the neighbouring participation with the anomeric centre. Then, 1, 2-cis-β-mannoside is formed after the selective removal of the acyl groups and C-2 configuration inversion, followed by the SN2 displacement reaction. The strategy of constructing β-taloside from β-galactoside is similar as above [22]. A synthetic approach to prepare disaccharide 6 (Scheme 2C) containing the mannosamine motif was developed starting from 5 (Scheme 2C). The free hydroxyl group of 5 was inverted to the desired configuration 6 by a triflate incorporation inversion strategy [41]. Recently, Adamo et al. synthesized the trisaccharide structure of the type 5 CPS repeating unit of Staphylococcus aureus, a major cause of nosocomial infections. As a main part of this trisaccharide, disaccharide 8 (Scheme 2C) with β-mannoside was synthesized through O-triflylation of disaccharide 7 (Scheme 2C) and SN2 displacement of the O-triflate with tetrabutylammonium (TBA) azide in 75% yield. This synthesized trisaccharide has been further linked to carrier proteins for vaccine investigations [42]. This strategy of constructing β-mannosides from β-glucosides through nucleophilic displacements via sulfonyl grouphas been previously reviewed in detail [38, 43].
Despite their low abundance, rare sugars hold great potential for medicine and drug applications [44]. Bacteria rare sugars are unique in pathogens, mostly absent in humans [45-49]. There are a variety of bacterial strains containing rare sugars on their cell surfaces, such as Shigella sonnei, Streptococcus mitis, Streptococcus pneumonia, Neisseria Meningitis and Pseudomonas aeruginosa[46, 50, 51]. Accordingly, bacterial CPS can be considered as candidate formulations for vaccine development [52-54]. However, the purification of carbohydrate compounds from bacteria in sufficient amounts is challenging. Hence, further efforts have been made to obtain rare sugars in good purities by chemical synthesis for glycoconjugate vaccine development.
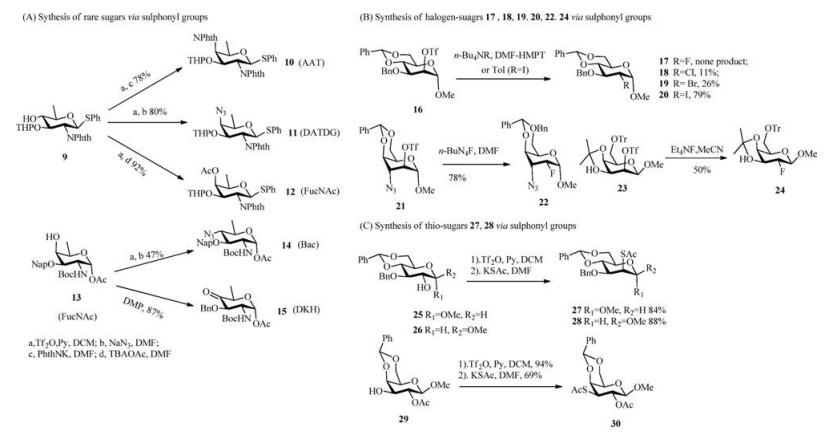
Recently, Ghosh et al. developed the chemical synthesis of bacterial rare sugar building blocks, such as 2-acetamido-4-amino-2, 4, 6-trideoxy-D-galactose (AAT) 10, 2, 4-diacetamido-2, 4, 6-trideoxy-D-galactose (DATDG) 11, N-acetylfucosamine (FucNAc) 12, from monosaccharide 9 (Scheme 3A), which can be prepared from common sugars, such as glucose and fucose, through deoxygenation at the C-6 position and azide incorporation inversion at C-4 and/or at C-2 [50, 51]. Meanwhile, Seeberger et al. developed a de novo route to synthesize Bac 14 and D-xylo-6-deoxy-4-ketohexos-amine (DKH) 15 from orthogonally protected FucNAc 13 by an inversion strategy via a triflate and oxidation [55]. A useful D-altro-pyranose derivative for synthesizing coriariin, found in C. japonica A, was synthesized from a D-mannopyranose derivative by steric inversion at the C-3 position in 62% yield [56]. Similarly, as a key synthon for synthesis of 1, 4-dideoxy-1, 4-imino-L-ribitol tetrahydroxy-LCB and α-galactosyl ceramide, allosamine derivative was prepared through epimerization of glucose O(3) —triflate with 74% yield [57].

|
Download:
|
| Scheme 3. Applied examples of inversion strategy via sulfonyl groups. (A) Sythesis of rare sugars via sulphonyl groups. (B) Synthesis of halogen-suagrs 17, 18, 19, 20, 22, 24 via sulphonyl groups. (C) Synthesis of thio-sugars 27, 28 via sulphonyl groups. | |
{kind=link}
Halogen sugars are carbohydrate derivatives in which one or more hydroxyls are replaced by a halogen atom(s). Karpiesiuk et al. studied the nucleophilic displacement of the 2-O-trifluorome-thylsulfonyl group on α-D-mannopyranoside with several charged halogen nucleophiles and found that the displacements proceeded complicatedly [58]. The best result was obtained for iodine substitution 20 in 79% yield, and fluorine substitution, as a poor nucleophile, to yield 17 was disfavoured for SN2 displacement (Scheme 3B). α-D-Idopyranoside 2-O-triflate 21 (Scheme 3B) and β-D-talopyranoside 2-O-triflate 23 (Scheme 3B) proceed smoothly with the corresponding inverted configurations 22 (Scheme 3B) and 24 (Scheme 3B) in 78% and 50% yields, respectively [59, 60]. Cumpstey et al. studied the nucleophilic displacement at α-D-glucopyranoside 25 and β-D-glucopyranoside 26 (Scheme 3C) with potassium thioacetate in DMF and obtained the corresponding 2-Smannosides 27 and 28 (Scheme 3C) in 84% and 88% yields [61]. The several SN2 displacements utilizing thioacetates as nucleophiles successfully proceeded with inverted configurations at the O-2, O-3 and O-4 positions of glucose [61, 62]. Thioacetate 30 (Scheme 3C) was obtained from galactose 29 (Scheme 3C) by a double inversion by Crich et al. [63].
3. Inversion via sequential oxidation/reductionOxidation followed by stereo selective reduction is the most reliable inversion method. First, the selectivity can be notably promoted by proceeding through LiH(sBu)3 (L-selectride) at a low temperature. Second, the stereoselectivity is predicted based on Cram's chelate model and depends on the neighbouring protecting groups. This is also a good alternative inversion method applied in carbohydrate synthesis
3.1. Mechanism of inversion via oxidation/reductionGenerally, the steps of sequential oxidation/reduction inverting the configuration of the hydroxyl group include chemical oxidation by Swern oxidation and stereoselectivity reduction with NaBH4, LiAlH4, L-selectride, etc. (Scheme 4A) [64, 65]. Although a variety of methods for this type of inversion are available, the most widely employed one is to use metal hydride reagents soluble in organic solvents such as, LiAlH4 and NaBH4. To complete the reduction, two hydrogen atoms are needed. The first hydrogen coming from a proton from the reducing agent is transferred from the boron or aluminium to the carbonyl group. The second one is taken from the solvent (when using NaBH4) or from added acid (when using LiAlH4). LiAlH4 is highly reactive and can produce H2 in polar solvents. Therefore, the reaction is performed in a polar aprotic solvent, such as diethyl ether or tetrahydrofuran (THF).

|
Download:
|
| Scheme 4. Mechanism and examples of oxidation/reduction inversion. (A) The general mechanism of oxidation/reduction inversion. (B) The Cram chelate model. (C) The benzyl ester chelating the metal favours the axial hydroxy group and the OBz group shows no chelation control. (D) 1, 3-Diaxial interactions direct the hydride to attack from the less hindered axial face. (E) The synthesis of 32, 34, 35 via oxidation/redunction inversion. (F) The preparation of β-mannoside likage of 39, 40, 41, 43, 45 via oxidation/redunction inversion. | |
{kind=link}
The selectivity can be predicted and explained based on Cram' chelate model, where the metal coordinates between the carbonyl and oxygen to form a staggered compound, and the red arrow indicates the hydride attack (Scheme 4B). The stereoselectivity of this reduction is normally influenced by two factors: (1) The benzyl esters that chelate the metal and favour attack of the hydride from the equatorial side (Scheme 4C); however, a benzoyl group (OBz) shows no chelation control; (2) 1, 3-Diaxial interactions that can direct the hydride to attack from the less hindered axial face (Scheme 4D).
3.2. The rules of inversion via oxidation/reductionSeveral rules can be summed up based on experimental evidence. The first rule is that the 2-ketosugar is normally proceeded with an equatorial hydroxyl in the terminal α-glycoside at the C(1) configuration. In addition, an axial form is usually obtained with a β-anomer [64-67]. The second one is that the 3-ketosugar can always proceed with an axial-hydroxyl configuration by a selected reduction reaction, which is normally facilitated by an adjacent equatorial alkoxyl group, such as OBn, p-methoxybenzyl group (OPMB), benzylidene, or isopropylidene, on the adjacent equatorial group and hampered under the presence of acyl groups, such as OBz, pivaloyl group (OPiv), trityl (OTr), or methoxymethyl group (OMOM), nearby [65, 68]. The third rule is that the 4-ketosugar normally proceeds with an axial hydroxyl when an equatorial benzyl group is nearby with equatorial OBn and OPMB protecting groups on the C-3 and C-6 nearby. When acyl or CH2 groups are nearby, the main product consists of an equatorial hydroxyl group [65, 69, 70].
3.3. Applications of inversion via oxidation/reductionThis method has been employed in inverting glucoside into mannoside [64, 65]. Cram's chelate model was developed for selective-reduction prediction [68]. Here, we focus on recent applications and innovations related to selected reduction with an introduction of inverted configuration. Very recently, a considerable development of selective oxidation was made by Chung et al., which is expected to promote the progress of stereoselective reduction [71, 72]. Bols et al., reported the syntheses of 6-deoxy-L-alloside, 6-deoxy-L-taloside, and 6-deoxy-L-altroside from L-fucose and L-rhamnose in high yields [65]. Using an oxidation/reduction inversion strategy, Boysen et al. synthesized the allosaminederived thioglycoside 32 (Scheme 4E) inverted from the C-3 of carbonyl-glucoside 31 (Scheme 4E) in 70% yield [57]. However, 33 (Scheme 4F) was reduced by L-selectride and NaBH4 leading to the C-3 axial-hydroxyl group of 34 (Scheme 4E) and the C-3 equatorial-hydroxyl group of 35 (Scheme 4E) in different proportions [72]. Apparently, L-selectride promoted the reduction selectivity. The borohydride reduction of 36 (Scheme 4F) was carried out in dichloromethane (DCM) and methanol, to give the β-mannoside derivative 39 (Scheme 4F) in excellent yield. Under the same conditions, 38 and 39 (Scheme 4F) were selectively reduced to afford b-mannoside 40 and 41 with 88% and 90% yield. β-Linkage is introduced by selective reduction at C-2 of 42 (Scheme 4F) and 44 (Scheme 4F) through the oxidation/reduction inversion strategy to obtain derivatives 43 (Scheme 4F) and 45 (Scheme 4F), which have been discussed extensively [69, 73, 74].
4. Inversion via triphenylphosphineThe substitution reaction of a primary or secondary hydroxyl by a trialkyl- or triarylphosphine and a dialkyl azodicarboxylate, known as the Mitsunobu reaction, was discovered by Oyo Mitsunobu in 1967 [75, 76]. This reaction has been successfully applied especially in stereoselective organic chemistry due to its mild reaction conditions, good stereoselectivity, and use of various proton reagents for different derivatives, such as inverted macrolides and amino- or halogen-modified organic species [77, 78]. It is generally agreed that a typical successful Mitsunobu reaction depends crucially not only on the pKa value of the pronucleophile, which must be approximately 11 or below, but also on the steric hindrance of the secondary hydroxyl group. This Mitsunobu reaction is particularly sensitive to the steric effect, which greatly limits its application on stereocongested carbocyclic alcohols [25, 65, 79-81]. Accordingly, compared to its numerous applications in organic chemistry, less Mitsunobu reactions have been used for inverting configurations in carbohydrate synthesis. However, the Mitsunobu reaction via PPh3 is irreplaceable to make a smooth inversion in some cases of carbohydrate synthesis [65].
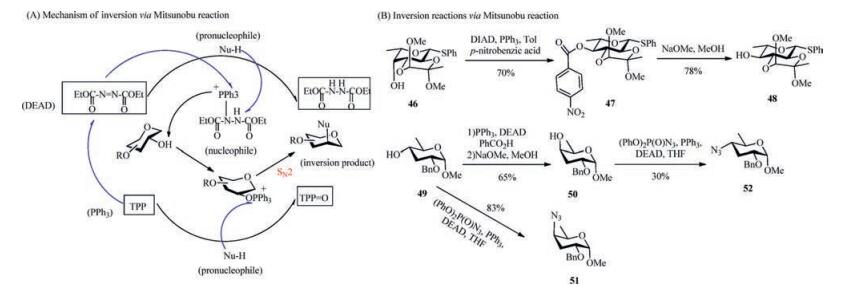
4.1. Mechanism of inversion via PPh3The mechanism of the Mitsunobu reaction with inversion by diethyl azodicarboxylate (DEAD) and PPh3 is depicted in Scheme 5A [77, 78]. The driving force of this process is the formation of a strong P=O bond. As seen here, this reaction proceeds with an inversion configuration at the secondary hydroxyl position. First, the lone electron pair of PPh3 attacks the N atom of DEAD to form a zwitterion, followed by combination with the pronucleophile to give the crucial nucleophile. Then, the nucleophile attacks the hydroxyl group, followed by inversion of the stereochemical configuration of the carbon centre by SN2 displacement. In this process, triphenylphosphine phosphine oxide is generated from PPh3, and a hydrazine derivative is derived from DEAD.

|
Download:
|
| Scheme 5. (A) Mechanism of Mitsunobu reaction. (B) Examples of the Mitsunobu reaction. | |
{kind=link}
4.2. Application of inversion via PPh3
Cléophax et al. reported the halogenation of carbohydrates with PPh3 [82]. Glucose derivatives with a free hydroxyl group on the C-4 position were first synthesized under microwave irradiation and in an oil bath. The inverted 2, 3, 4-O-benzyl-4-deoxy-chloro-galactose derivative and 2, 3, 4-O-benzyl-4-deoxy-bromo-galactose derivative were then obtained in 85% and 65% yields, respectively. An inversion strategy via PPh3 was applied to the selective conversion of a 2-OBz-D-fructopyranose derivative with free cis-diols into iodohydrins in 93% conversion yield. In addition, this suggested a trend to form trans-diols using the Mitsunobu reaction [65, 83]. Pedersen et al. performed a Mitsunobu inversion reaction from 6-deoxy-L-galactopyranoside 46 (Scheme 5B), followed by Zemplén diacylation, to obtain 6-deoxy-L-glucopyranoside 48 (Scheme 5B) in 70% yield, which did not proceed smoothly by inversion via a sulfonyl group [37, 65, 84]. Additionally, 6-deoxy-Lidoside was synthesized from 6-deoxyl-L-altroside in a very high yield of 98% by a Mitsunobun reaction, but this procedure was not successful using an oxidation/reduction route [65]. An inversion strategy by the Mitsunobun reaction was used three times for the preparation of 3, 6-didexy-hexoses paratose 49 (Scheme 5B) and abequose 50 (Scheme 5B), as well as their azide-substituted analogues 51 (Scheme 5B) and 52 (Scheme 5B). The azide introduction was carried out through a Mitsunobun azide substitution reaction with diphenyl phosphoryl azide, PPh3, and DEAD, providing the responding converted azide analogues 51 and 52 in 83% and 30% yields, respectively. Actually, the benzylprotected 3, 6-dideoxyhexose 50 was inverted from 49 by a Mitsunobun reaction, followed by treatment with sodium methoxide and methanol, in 65% yield [85].
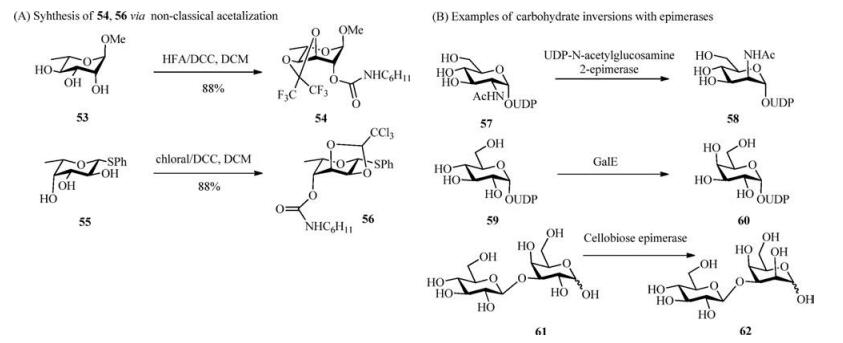
5. Inversion via other methodsEpimerization by non-classical acetalization involving a threecomponent reaction is a highly attractive route, as the epimerization occurs at the same time as the installation of orthogonal protecting groups and allows for the use of unprotected triols or tetrols for the stereoselective epimerization of carbohydrates. This acetalization/epimerization strategy was developed by Miethchen et al. [86-89]. A particular advantage of the one-pot acetalization/epimerization method by chloral/N, N'-dicyclohexylcarbodiimide (DCC) is the access to rare natural or non-natural sugars from common monosaccharides, for example, gulose from galactose, altrose from mannose, and tagatose from fructose [90-92]. Sugar epimerization via acetalization by chloral/DCC or hexafluoroacetone (HFA)/DCC has been previously reviewed [93]. Miethchen et al. developed a non-conventional acetalization of methyl α-L-rhamno pyranoside towards a highly stereoselective epimerization compound in a one-pot procedure. Using this HFA/DCC-induced inversion strategy, the L-altrose derivative 54 (Scheme 6A) was inverted from the L-rhmnose derivative 53 (Scheme 6A) in the presence of HFA, DCC and DCM [86]. Under similar conditions, the L-fucose derivative 55 (Scheme 6A) was inverted to afford the Lgulose derivative 56 (Scheme 6A) in 88% yield [65].

|
Download:
|
| Scheme 6. Carbohydrate inversions by non-classical acetalization and different epimerases. | |
{kind=link}
Enzymatic-induced inversions involving carbohydrate epimerases, such as epimerases and racemases, catalysing an inversion of a stereogenic centre have been utilized to obtain a converted configuration in a sugar [94-98]. Various epimerases have been employed in the conversion of rare sugars from cheap and widely available substrates [44]. For example, D-psicose 3-epimerase and D-tagatose 3-epimerase were utilized in converting D-fructose to Dpsicose, which is an ultra-low-energy monosaccharide sugar potentially used as a food sweetener to combat hyperglycaemia, diabetes and obesity [95, 99, 100]. Many other rare sugars have also been synthesized with different epimerases, such as epilactose and D-tagatose, utilizing their prebiotic properties; GalNAc, lacto-N-biose, and galacto-N-biose, making the core structures of functional sugar chains; and D-talose, D-allose, L-xylose, L-ribose, L-galactose, and L-gulose, used as building blocks to synthesize antiviral and anticancer drugs [44, 101-108]. Tanner et al. used the bifunctional UDP-N-acetylglucosamine 2-epimerase, which catalyses the biosynthesis of sialic acids in mammals, to convert UDPGlcNAc 57 (Scheme 6B) into ManNAc 58 (Scheme 6B) and UDP [109]. Ketohexose 3-epimerase shows a wide range of substrate specificity in catalysing at the C-3 position, such as interconverting D-tagatose with D-sorbose, D-fructose with D-psicose, D-ribulose with d-xylulose and d-ribulose with D-xylulose [110, 111]. UDPgalactose 4-epimerase (GalE) is essential in galactose metabolism and has been used to catalyse the inversion of glucose 59 (Scheme 6B) to galactose 60 (Scheme 6B) [112, 113]. Cellobiose epimerase from Ruminococcus albus has been used in interconverting β-D-glucopyranosyl-(1→4)-D-glucose 61 (Scheme 6B) and β-Dglucopyranosyl-(1→4)-D-mannose 62 (Scheme 6B) [114].
6. Summary and outlookVarious inversion synthetic methodologies, covering their mechanisms and applications, have been reviewed, as they represent powerful tools to synthesize corresponding inverted configurations. These inversion strategies, including inversion via sulfonyl groups, oxidation/selective reduction, PPh3, epimerization by non-classical acetalization and enzymatic-induced inversions, have been widely used to synthesize amino sugars and other deoxyl sugars. Rare sugars can be abundantly obtained by these inversion strategies from common sugars. In addition to introducing functionalities and abundantly synthesizing rare sugars, inversion methods are also used in constructing the β-configurations in glycosylation.
Inversion strategies for the corresponding inverted configurations are essential in carbohydrate synthesis and vaccine development. A promising outlook for developing inversion variations and new epimerization methods is provided from these methods mentioned above with their respective advantages and limits. It is expected that, with this comprehensive review on the different mechanisms, rules and applications of inversion methods, the community can avoid possible pitfalls and effectively plan future routes to target sugar derivatives.
AcknowledgementsWe thank the National Natural Science Foundation of China (No. 21302068), the Natural Science Foundation of Jiangsu Province, China (No. BK20150140), the Fundamental Research Funds for the Central Universities (No. JUSRP51712B) and the Public Health Research Center at Jiangnan University (No. JUPH201502).
| [1] |
A. Varki, Glycobiology 3(1993) 97-130. DOI:10.1093/glycob/3.2.97 |
| [2] |
J.B. Lowe, J.D. Marth, Annu. Rev. Biochem. 72(2003) 643-691. DOI:10.1146/annurev.biochem.72.121801.161809 |
| [3] |
M. Trent, C.M. Stead, A.X. Tran, et al., J. Endotoxin. Res. 12(2006) 205-223. |
| [4] |
H. Sahly, Y. Keisari, E.C. Crouch, et al., Infect. Immun. 76(2008) 1322-1332. DOI:10.1128/IAI.00910-07 |
| [5] |
C. Weidenmaier, A. Peschel, Nat. Rev. Microbiol. 6(2008) 276-287. DOI:10.1038/nrmicro1861 |
| [6] |
F. Broecker, J. Hanske, C.E. Martin, et al., Nat. Commun. 7(2016) 11224. DOI:10.1038/ncomms11224 |
| [7] |
H. Liu, D.J. Irvine, Bioconjugate Chem. 26(2015) 791-801. DOI:10.1021/acs.bioconjchem.5b00103 |
| [8] |
T.J. Boltje, T. Buskas, G. Boons, Nat. Chem. 1(2009) 611-622. DOI:10.1038/nchem.399 |
| [9] |
R.M.F.V. Der Put, T.H. Kim, C. Guerreiro, et al., Bioconjugate Chem. 27(2016) 883-892. DOI:10.1021/acs.bioconjchem.5b00617 |
| [10] |
M.A. Monteiro, Z. Ma, L. Bertolo, et al., Expert. Rev. Vaccines 12(2014) 421-431. |
| [11] |
C.E. Martin, M. Weishaupt, P.H. Seeberger, Chem. Commun. 47(2011) 10260-10262. DOI:10.1039/c1cc13614c |
| [12] |
(a) H. Dong, Efficient Carbohydrate Synthesis by Controlled Inversion Strategies, Licentaite Thesis, Kungliga Tekniska högskolan (Royal Institute of Technology), 2006; (b) H. Dong, Efficient Carbohydrate Synthesis by Intra-and Supramolecular Control, Doctoral Thesis, Kungliga Tekniska högskolan (Royal Institute of Technology), 2008. |
| [13] |
K.J. Hale, L. Hough, S. Manaviazar, et al., Org. Lett. 16(2014) 4838-4841. DOI:10.1021/ol502193j |
| [14] |
A.C. Richardson, Carbohydr. Res. 10(1969) 395-402. DOI:10.1016/S0008-6215(00)80900-3 |
| [15] |
R. Lattrell, G. Lohaus, Liebigs Ann. Chem. 1974(1974) 901-920. DOI:10.1002/(ISSN)1099-0690 |
| [16] |
R. Albert, K. Dax, R.W. Link, et al., Carbohydr. Res. 118(1983) C5-C6. DOI:10.1016/0008-6215(83)88062-8 |
| [17] |
H. Dong, Pei Zhichao, O. Ramström, J. Org. Chem. 71(2006) 3306-3309. DOI:10.1021/jo052662i |
| [18] |
S. Knapp, A.B. Naughton, C. Jaramillo, et al., J. Org. Chem. 57(1992) 7328-7334. DOI:10.1021/jo00052a058 |
| [19] |
G. Dijkstra, W. Kruizinga, R.M. Kellogg, J. Org. Chem. 52(1987) 4230-4234. DOI:10.1021/jo00228a015 |
| [20] |
M. Miljković, Nucleophilic Displacement and the Neighboring Group Participation, Carbohydrates[M]. New York: Springer, 2010, pp. 169-190.
|
| [21] |
H. Dong, M. Rahm, N. Thota, et al., Org. Biomol. Chem. 11(2013) 648-653. DOI:10.1039/C2OB26980E |
| [22] |
H. Dong, Z.C. Pei, M. Angelin, et al., J. Org. Chem. 72(2007) 3694-3701. DOI:10.1021/jo062643o |
| [23] |
Z.C. Pei, H. Dong, R. Caraballo, et al., Eur. J. Org. Chem.(2007), 4927-4934. |
| [24] |
D.S. Noyce, J.A. Virgilio, J. Org. Chem. 37(1972) 2643-2647. DOI:10.1021/jo00982a001 |
| [25] |
H. Ochiai, T. Niwa, T. Hosoya, Org. Lett. 18(2016) 5982. DOI:10.1021/acs.orglett.6b02675 |
| [26] |
R.W. Binkley, J. Org. Chem. 56(1991) 3892-3896. DOI:10.1021/jo00012a020 |
| [27] |
D.R. Dalton, R.C.S. Jr, D.G. Jones, Tetrahedron 26(1970) 575-581. DOI:10.1016/S0040-4020(01)97850-0 |
| [28] |
J. Muzart, Tetrahedron 65(2009) 8313-8323. DOI:10.1016/j.tet.2009.06.091 |
| [29] |
Y. Cai, C.C. Ling, D.R. Bundle, J. Org. Chem. 74(2009) 580-589. DOI:10.1021/jo801927k |
| [30] |
G.W.J. Fleet, M.J. Gough, T.K.M. Shing, Tetrahedron Lett. 25(1984) 4029-4032. DOI:10.1016/0040-4039(84)80058-1 |
| [31] |
J. Vesely, A. Rohlenova, M. Dzoganova, et al., Synthesis(2006), 699-705. |
| [32] |
A. Banerjee, S.K. Ghosh, Mol. Cell. Biochem. 253(2003) 179-190. DOI:10.1023/A:1026058311857 |
| [33] |
G.A. Ellestad, D.B. Cosulich, R.W. Broschard, et al., J. Am. Chem. Soc. 100(1978) 2515-2524. DOI:10.1021/ja00476a041 |
| [34] |
D. Leonori, P.H. Seeberger, Org. Lett. 14(2012) 4954-4957. DOI:10.1021/ol3023227 |
| [35] |
C.T. Oberg, A. Noresson, T. Delaine, et al., Carbohydr. Res. 344(2009) 1282-1284. DOI:10.1016/j.carres.2009.05.005 |
| [36] |
B. Dhakal, S. Buda, D. Crich, J. Org. Chem. 81(2016) 10617-10630. DOI:10.1021/acs.joc.6b02221 |
| [37] |
S.R. Sanapala, S.S. Kulkarni, J. Am. Chem. Soc. 138(2016) 4938-4947. DOI:10.1021/jacs.6b01823 |
| [38] |
E.S.H.E. Ashry, N. Rashed, E.S.I. Ibrahim, Curr. Org. Synth. 37(2006) 175-213. |
| [39] |
W. Günther, H. Kunz, Carbohydr. Res. 228(1992) 217. DOI:10.1016/S0008-6215(00)90561-5 |
| [40] |
M. Miljković, M. Gligorijević, D. Glisin, J. Org. Chem. 39(1974) 3223-3226. DOI:10.1021/jo00936a009 |
| [41] |
M.A. Oberli, P. Bindschädler, D.B. Werz, et al., Org. Lett. 10(2008) 905-908. DOI:10.1021/ol7030262 |
| [42] |
E. Danieli, D. Proietti, G. Brogioni, et al., Bioorg. Med. Chem. 20(2012) 6403-6415. DOI:10.1016/j.bmc.2012.08.048 |
| [43] |
S. David, A. Malleron, C. Dini, Carbohydr. Res. 188(1989) 193-200. DOI:10.1016/0008-6215(89)84070-4 |
| [44] |
K. Beerens, T. Desmet, W. Soetaert, J. Ind. Microbiol. Biotechnol. 39(2012) 823-834. DOI:10.1007/s10295-012-1089-x |
| [45] |
S.A. Longwell, D.H. Dube, Curr. Opin. Chem. Biol. 17(2013) 41. DOI:10.1016/j.cbpa.2012.12.006 |
| [46] |
D.H. Dube, K. Champasa, B. Wang, Chem. Commun. 47(2011) 87. DOI:10.1039/C0CC01557A |
| [47] |
A. Adibekian, P. Stallforth, M.L. Hecht, et al., Chem. Sci. 2(2010) 337-344. |
| [48] |
L. Kenne, B. Lindberg, K. Petersson, et al., Carbohydr. Res. 78(1980) 119-126. DOI:10.1016/S0008-6215(00)83665-4 |
| [49] |
H. Baumann, A.O. Tzianabos, J.R. Brisson, et al., Biochemistry 31(1992) 4081-4089. DOI:10.1021/bi00131a026 |
| [50] |
A. Chaudhury, R. Ghosh, Org. Biomol. Chem.(2017), 1444-1452. |
| [51] |
M. Emmadi, S.S. Kulkarni, Nat. Prod. Rep. 31(2014) 870-879. DOI:10.1039/C4NP00003J |
| [52] |
L. Morelli, L. Poletti, L. Lay, Eur. J. Org. Chem.(2011), 5723-5777. |
| [53] |
A. Fernández-Tejada, F.J. Cañada, J. Jiménez-Barbero, Chem. Eur. J. 21(2015) 10616-10628. DOI:10.1002/chem.v21.30 |
| [54] |
P.H. Seeberger, D.B. Werz, Nature 446(2007) 1046-1051. DOI:10.1038/nature05819 |
| [55] |
R. Pragani, P.H. Seeberger, J. Am. Chem. Soc. 133(2011) 102-107. DOI:10.1021/ja1087375 |
| [56] |
S. Matsuda, T. Yamanoi, M. Watanabe, Tetrahedron 64(2008) 8082-8088. DOI:10.1016/j.tet.2008.06.068 |
| [57] |
S.Y. Luo, S.S. Kulkarni, C.H. Chou, et al., J. Org Chem. 71(2006) 1226-1229. DOI:10.1021/jo051518u |
| [58] |
W. Karpiesiuk, A. Banaszek, A. Zamojski, Carbohydr. Res. 186(1989) 156-162. DOI:10.1016/0008-6215(89)84013-3 |
| [59] |
L.H.B. Baptistella, A.J. Marsaioli, P.M. Imamura, et al., Carbohydr. Res. 152(1986) 310-315. DOI:10.1016/S0008-6215(00)90313-6 |
| [60] |
T. Haradahira, M. Maeda, Y. Yano, et al., Chem. Pharm. Bull. 32(1984) 3317-3319. DOI:10.1248/cpb.32.3317 |
| [61] |
I. Cumpstey, C. Ramstadius, T. Akhtar, et al., Eur. J. Org. Chem.(2010), 1951-1970. |
| [62] |
I. Cumpstey, D.S. Alonzi, T.D. Butters, Carbohydr. Res. 344(2009) 454-459. DOI:10.1016/j.carres.2008.12.023 |
| [63] |
A. Noel, B. Delpech, D. Crich, Organ. Lett. 14(2012) 4138-4141. DOI:10.1021/ol301779e |
| [64] |
C.T. Chang, Y. Hui, B. Elchert, Tetrahedron Lett. 42(2001) 7019-7023. DOI:10.1016/S0040-4039(01)01472-1 |
| [65] |
T.G. Frihed, C.M. Pedersen, M. Bols, Eur. J. Org. Chem.(2014), 7924-7939. |
| [66] |
D.J. Cram, K.R. Kopecky, J. Am. Chem. Soc. 81(1959) 2748-2755. DOI:10.1021/ja01520a036 |
| [67] |
F.W. Lichtenthaler, T. Schneideradams, J. Org. Chem. 59(1994) 6728-6734. DOI:10.1021/jo00101a035 |
| [68] |
A. Mengel, O. Reiser, Chem. Rev. 99(1999) 1191-1224. DOI:10.1021/cr980379w |
| [69] |
H.C. Brown, S. Krishnamurthy, J. Am. Chem. Soc. 94(1972) 7159-7161. DOI:10.1021/ja00775a053 |
| [70] |
M.R. Dhawale, W.A. Szarek, G.W. Hay, et al., Carbohydr. Res. 155(1986) 262-265. DOI:10.1016/S0008-6215(00)90156-3 |
| [71] |
K. Chung, R.M. Waymouth, ACS Catal. 6(2016) 4653-4659. DOI:10.1021/acscatal.6b01501 |
| [72] |
V.R. Jumde, N.N. Eisink, M.D. Witte, et al., J. Org. Chem. 81(2016) 11439-11443. DOI:10.1021/acs.joc.6b02074 |
| [73] |
E.S.H.E. Ashry, N. Rashed, E.S.I. Ibrahim, Tetrahedron 64(2008) 10631-10648. DOI:10.1016/j.tet.2008.09.001 |
| [74] |
F.W. Lichtenthaler, T. Metz, Eur. J. Org. Chem.(2003), 3081-3093. |
| [75] |
O. Mitsunobu, M. Yamada, Bull. Chem. Soc. Jpn. 40(1967) 2380-2382. DOI:10.1246/bcsj.40.2380 |
| [76] |
O. Mitsunobu, M. Yamada, T. Mukaiyama, Bull. Chem. Soc. Jpn. 40(1967) 935-939. DOI:10.1246/bcsj.40.935 |
| [77] |
K.C.K. Swamy, N.N.B. Kumar, E. Balaraman, et al., Chem. Rev. 109(2009) 2551-2651. DOI:10.1021/cr800278z |
| [78] |
S. Fletcher, Org. Chem. Front. 2(2015) 739-752. DOI:10.1039/C5QO00016E |
| [79] |
B.K. Shull, T. Sakai, J.B. Nichols, et al., J. Org. Chem. 62(1997) 8294-8303. DOI:10.1021/jo9615155 |
| [80] |
C.A. Hoeger, A.D. Johnston, W.H. Okamura, J. Am. Chem. Soc. 109(1987) 4690-4698. DOI:10.1021/ja00249a035 |
| [81] |
G. Wang, J.R. Ella-Menye, M.M. St, et al., Org. Lett. 10(2008) 4203-4206. DOI:10.1021/ol801316f |
| [82] |
C. Limousin, A. Olesker, J. Cléophax, et al., Carbohydr. Res. 312(1998) 23-31. DOI:10.1016/S0008-6215(98)00224-9 |
| [83] |
A.C. Simao, A. Tatibouet, A.P. Rauter, et al., Tetrahedron Lett. 51(2010) 4602-4604. DOI:10.1016/j.tetlet.2010.06.107 |
| [84] |
T.G. Frihed, M. Bols, C.M. Pedersen, Chem. Rev. 115(2015) 3615-3676. DOI:10.1021/acs.chemrev.5b00104 |
| [85] |
C.W.T. Chang, T. Clark, M. Ngaara, Tetrahedron Lett. 42(2001) 6797-6801. DOI:10.1016/S0040-4039(01)01402-2 |
| [86] |
R. Miethchen, D. Rentsch, M. Michalik, Eur. J. Org. Chem.(1994), 219-222. |
| [87] |
M. Frank, R. Miethchen, H. Reinke, Eur. J. Org. Chem.(1999), 1259-1263. |
| [88] |
R. Miethchen, D. Rentsch, M. Frank, J. Carbohydr. Chem. 15(1996) 15-31. DOI:10.1080/07328309608005421 |
| [89] |
C. Hager, R. Miethchen, H. Reinke, Synthesis(2000), 226-232. |
| [90] |
R. Miethchen, M.L.A. Frank, D. Rentsch, Carbohydr. Res. 281(1996) 61-68. DOI:10.1016/0008-6215(95)00338-X |
| [91] |
D. Rentsch, R. Miethchen, Carbohydr. Res. 239(1996) 139-145. |
| [92] |
M. Frank, R. Miethchen, D. Degenring, Carbohydr. Res. 318(1999) 167-170. DOI:10.1016/S0008-6215(99)00090-7 |
| [93] |
R. Miethchen, J. Carbohydr. Chem. 22(2004) 801-825. |
| [94] |
S. Van Overtveldt, T. Verhaeghe, H. Joosten, et al., Biotechnol. Adv. 33(2015) 1814-1828. DOI:10.1016/j.biotechadv.2015.10.010 |
| [95] |
J. Samuel, M.E. Tanner, Nat. Prod. Rep. 19(2002) 261-277. DOI:10.1039/b100492l |
| [96] |
S.-M. Shin, J.M. Choi, E.d. Luccio, et al., Arch. Biochem. Biophys. 585(2015) 39-51. DOI:10.1016/j.abb.2015.08.025 |
| [97] |
K. Beerens, W. Soetaert, T. Desmet, Carbohydr. Res. 414(2015) 8-14. DOI:10.1016/j.carres.2015.06.006 |
| [98] |
L. Zhang, M.M. Muthana, H. Yu, et al., Carbohydr. Res. 419(2016) 18-28. DOI:10.1016/j.carres.2015.10.016 |
| [99] |
K. Kim, H. Kim, D. Oh, et al., J. Mol. Biol. 361(2006) 920-931. DOI:10.1016/j.jmb.2006.06.069 |
| [100] |
H. Yoshida, M. Yamada, T. Nishitani, et al., J. Mol. Biol. 374(2007) 443-453. DOI:10.1016/j.jmb.2007.09.033 |
| [101] |
M. Krewinkel, J. Kaiser, M. Merz, et al., J. Dairy Sci. 98(2015) 3665-3678. DOI:10.3168/jds.2015-9411 |
| [102] |
J. Watanabe, M. Nishimukai, H. Taguchi, et al., J. Dairy Sci. 91(2008) 4518-4526. DOI:10.3168/jds.2008-1367 |
| [103] |
M.L. Sanz, G.R. Gibson, R.A. Rastall, J. Agric. Food Chem. 53(2005) 5192-5199. DOI:10.1021/jf050276w |
| [104] |
Z. Li, Y. Gao, H. Nakanishi, et al., Beilstein J. Org. Chem. 9(2013) 2434-2445. DOI:10.3762/bjoc.9.281 |
| [105] |
R. Woodyer, T.N. Christ, K.A. Deweese, Carbohydr. Res. 345(2010) 363-368. DOI:10.1016/j.carres.2009.11.023 |
| [106] |
D. Rao, P. Gullapalli, A. Yoshihara, et al., J. Biosci. Bioeng. 106(2008) 473-480. DOI:10.1263/jbb.106.473 |
| [107] |
K. Inoue, M. Nishimoto, M. Kitaoka, Carbohydr. Res. 346(2011) 2432-2436. DOI:10.1016/j.carres.2011.08.032 |
| [108] |
M. Nishimoto, M. Kitaoka, Carbohydr. Res. 344(2009) 2573-2576. DOI:10.1016/j.carres.2009.09.031 |
| [109] |
W.K. Chou, S. Hinderlich, W. Reutter, et al., J. Am. Chem. Soc. 125(2003) 2455-2461. DOI:10.1021/ja021309g |
| [110] |
H. Itoh, H. Okaya, A.R. Khan, et al., Biosci. Biotechnol. Biochem. 58(1994) 2168-2171. DOI:10.1271/bbb.58.2168 |
| [111] |
K. Izumori, A.R. Khan, H. Okaya, et al., Biosci. Biotechnol. Biochem. 57(1993) 1037-1039. DOI:10.1271/bbb.57.1037 |
| [112] |
J.B. Thoden, P.A. Frey, H.M. Holden, Protein Sci. 5(1996) 2149-2161. DOI:10.1002/pro.v5:11 |
| [113] |
P.A. Frey, A.D. Hegeman, Acc. Chem. Res. 46(2013) 1417-1426. DOI:10.1021/ar300246k |
| [114] |
T.R. Tyler, J.M. Leatherwood, Arch. Biochem. Biophys. 119(1967) 363-367. DOI:10.1016/0003-9861(67)90466-3 |