 2017, Vol. 28
2017, Vol. 28 



b Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China;
c State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116012, China
Fluorescent labeling of interested protein in vivo plays an increasingly important role in studying the dynamics, localization and functions of proteins in living cells. Fusing the protein of interest (POI) with fluorescent proteins (FPs) is the traditional method utilized by biologists [1-3]. However, the application of FPs in vivo is sometimes limited by their relatively large size, the narrow spectral range, and considerable proneness to photobleaching after prolonged excitation. In contrast, small molecule fluorophores display the characteristic of emissions spanning the color spectrum and excellent photochemical or photophysical properties (i.e., high quantum yield, good photostability, ability to photoswitch, etc.), which are much advantageous over FPs [4, 5]. But an inherent disadvantage of organic fluorophores is that they cannot be accurately located and quantified to POI as genetically encoded FPs. To overcome this limitation, protein-tags labeling technique has been greatly developed over the past decade. In this approach, protein tags were fused to the POI in vivo and organic small molecule fluorophores spontaneously react with their protein tags through highly specific chemical reactions, which are so-called bioorthogonal reactions [6, 7].
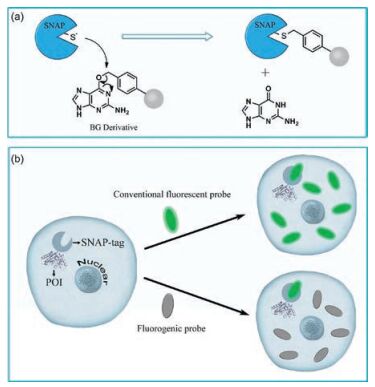
To date, various protein tags have been developed to label POI in living systems, such as HaloTag [8, 9], SNAP-tag [10, 11], TMP-tag [12], PYP-tag [13-15], BL-tag [16, 17], and so on. Among them, SNAP-tag, pioneered by Johnsson and co-workers, has been recognized as one of the most prominent fusion tags [18]. SNAP-tag is a 20-kDa engineered mutant of the human repair protein O6-alkylguanine-DNA alkyltransferase (hAGT), which rapidly and specifically reacts with para-substituted O6-benzylguanine (BG) derivatives by transferring the substituted benzyl group to its reactive site through a nucleophilic substitution reaction while releasing free guanine (Fig. 1a) [19, 20]. Compared with others, the advantages of SNAP-tags are their broad applications in vivo and in vitro in different genetically modified organisms like bacteria (Escherichia coli), yeast, and mammalian cells, at a variety of substrates which can be easily generated with excellent reaction rate [21, 22]. In consideration of potential applications, one tag that is comparable to the SNAP-tag is the Halo-tag. However, it is hard to generate fluorogenic probes for Halo-Tags ascribing to the introduction of fluorophores into its substrates. In living cells, SNAP-tag was firstly expressed as a fusion to the targeted protein domain, and then a fluorophore-derived BG compound was loaded to tag SNAP with the fluorophore and subsequently achieve the living cell imaging of POI by fluorescence microscopy. Nowadays, various fluorescent substrates have been designed to react with SNAP-tag and applied widely in drug monitoring, protein-protein interactions, fluorescent sensors and super-resolution fluorescence imaging [23-25]. While these probes displayed a great potential to study proteins, the step of washing out unreacted probes is required before imaging. Besides the tedious and time-consuming process, this wash-out requirement may potentially limit their application in real-time monitoring of molecular events such as receptor-ligand binding, endocytosis, trafficking and so on. In addition, to wash out unreacted probes completely are difficult because of the accumulation of the synthetic probes in various organelles [26-30].

|
Download:
|
| Fig. 1. (a) Reaction mechanism of SNAP-tag with BG derivative. (b) Protein labeling system with SNAP-tag and fluorogenic probe for live-cell imaging without and with turn-on effect, respectively | |
{kind=link}
To overcome this limitation, fluorogenic probes have been designed (Fig. 1b). Fluorogenic probes exhibit no fluorescence and display a selective turn-on fluorescence response when they bind to SNAP-tags. Thus, much higher signal-to-noise ratios than conventional fluorescent probes would be achieved, which minimize the fluorescence background caused by unreacted or nonspecifically bound probes and allow fluorogenic probes for direct imaging in living cells without wash-out steps. In other words, free probes are not fluorescent or only weakly fluorescent in this fluorogenic system, while SNAP-tag expressed in the living cells are detected quickly and selectively without washing the cells after being labeled with the probes. Here, we focus on the design strategies of fluorogenic probes and the applications in wash-free protein labeling in living cells.
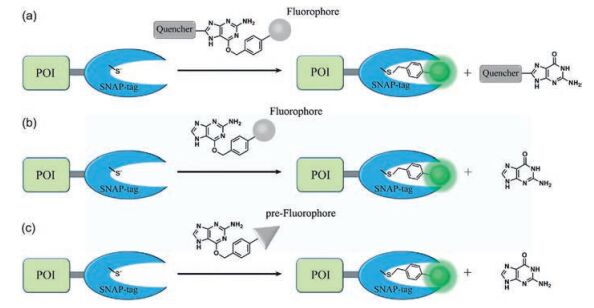
2. Fluorogenic probesA typical fluorogenic probe for SNAP-tag contains a BG group linked to a fluorophore which translates the reaction between SNAP and BG group into a significant fluorescence change. Therefore, a fluorogenic probe must meet two basic requirements: fluorescence signals capable of avoiding background interference and the high reaction-induced fluorescence signal selectivity. The signal of fluorogenic probes is usually measured as a change in fluorescence intensity, fluorescence lifetime, or a shift of fluorescence wavelength. As shown in Fig. 2, three strategies at present are usually applied to design fluorogenic probes including releasing fluorescence quenchers, utilizing environmental sensitivity of fluorophores and generating fluorophores in situ.

|
Download:
|
| Fig. 2. Strategies of designing SNAP-tag fluorogenic probes with turn-on switch. (a) Release of fluorescence quencher; (b) environmental sensitivity of fluorophores; and (c) in situ fluorophore generation. | |
{kind=link}
2.1. Release of fluorescence quencher
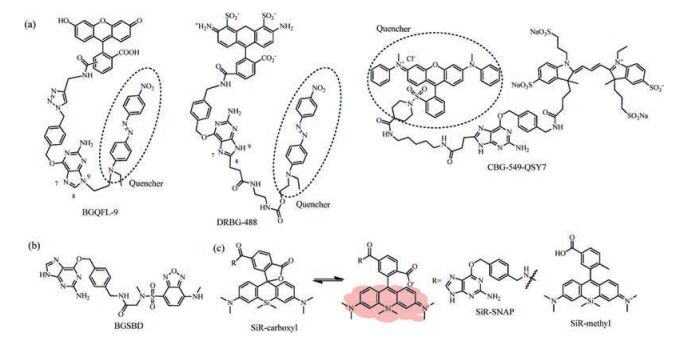
The SNAP-tag labeling reaction occurs by the nucleophilic attack of reactive Cys to the benzyl carbon of the probe and the simultaneous formation of a stable thioether bond between the probe and protein, resulting in release of the guanine group (Fig. 1a). Since guanine is an electron-rich group and well-known to quench the fluorescence of certain dyes by photo-induced electron transfer (PET), Herten and coworkers synthesized 21 different BG-dye conjugates to study intramolecular fluorescence quenching by guanine [31]. And nine of them showed potentials in living cell imaging. Although this method is simple and avoids complicated modifying of substrates, but the quenching efficiency is not satisfactory in real-time imaging experiments. Thus, strong fluorescence quenchers were incorporated into the guanine moiety (Fig. 2a). Yao et al. synthesized a fluorogenic probe, BGQFL-9 (Fig. 3a), with Disperse Red 1 (DR1) as a fluorescence quencher since the absorption spectrum of DR1 overlaps substantially with the emission spectra of fluorescein [32]. DR1 is modified at the N9 position of the guanine moiety, and the probe showed almost no fluorescence. At the same time, Urano et al. reported that modification at either N9 or C8 position of O6-benzylguanine derivatives would avoid the influence of substrate recognition of the enzyme. And the reaction rates of modification at C8 position was superior from the viewpoint of fast labeling of SNAP-tag (C8-carboxyethyl-BG, k ~ 5 ×103 L mol-1 s-1, N9-methoxyethanol-BG, k < 1 L mol-1 s-1) [33]. Based on a similar approach, Sun et al. developed a fluorogenic probe based on FRET mechanism, CBG-549-QSY7 (Fig. 3a), with QSY7 as the fluorescence quencher bound to C8 position of the guanine moiety [34]. The quenching efficiency was greater than 98%. The authors conclude that the quenching efficiency of the fluorogenic substrates was a result of both the FRET process and quenching ability of guanine. In addition, a series of fluorogenic substrates with different fluorescence colors were created using fluorophore/quencher pairs from commercially available probes. Since the quenching efficiency was greater than 95%, the labeling reaction occurred slowly with the second-order rate constant between the SNAP-tag and probes ranged from 0.152 L mol-1 s-1 to 0.831 ×103 L mol-1 s-1. The reaction rates of the fluorogenic probes are much lower than the substrates without the modification at the guanine moiety (4.5-12.2 ×103 L mol-1 s-1). However, they will be useful for multicolor imaging analysis of protein localization.

|
Download:
|
| Fig. 3. Chemical structure of fluorogenic probes for SNAP-tag. (a) Probes based on quencher release; (b) probes based on fluorophore's environmental sensitivity; and (c) probes based on siliconrhodamine (SiR) fluorophore. And the equilibrium of carboxylated SiR between its nonfluorescent spirolactone and its fluorescent zwitterion. | |
{kind=link}
2.2. Environment sensitivity of fluorophore
Fluorescent dyes with large solvatochromism are a wellestablished class of environment-sensitive fluorophore. They exhibit weak fluorescence in polar solvents but strong emissions in low-polarity solvents and viscous environments [35]. Representative fluorophores include Nile red, NBD, and naphthalimide, which are also effective probes for protein polarity. According to the analysis of the crystal structure of SNAP-tag, the BG-binding activity center of the SNAP-tag is a hydrophobic narrow cavity. Therefore, the substrate with small size and environment-sensitive fluorophore moiety could fit into the low-polar pocket of SNAP-tag and a covalent bond was shown to form between the probe and SNAP-tag followed by turn-on fluorescence (Fig. 2b). Tan et al. developed a fluorogenic probe, BGSBD, based on an environmentsensitive fluorophore 4-sulfamonyl-7-aminobenzoxadiazole (SBD), which is more sensitive to changes in polarity and the hydrogen bonding ability of a solvent (Fig. 3b) [36]. The fluorogenic substrate can rapidly label SNAP-tag proteins with fluorescence increase of 280-fold. And the second-order rate constant between BGSBD and SNAP-tag was determined to be 7.2 ×103 L mol-1 s-1. It is a great SNAP-tag fluorogenic probe to different cellular compartments of living cells without a wash-out process. Furthermore, BGSBD analogs were designed for the quantitative determination of two biologically important targets, nitroreductase and hydrogensulfide, in blood plasma [37]. Our group also created a SNAP-tag fluorogenic probe with 1, 8-naphthalimide. The probe based on both environmental sensitivity of 1, 8-naphthalimide and fluorescence quenching effect of guanine. The secondorder rate constant was about 2.1 ×103 L mol-1 s-1 and the fluorescence quantum yield displayed significantly enhancement after the reaction with SNAP-tag. This probe can cross cell membrane and rapidly label intracellular proteins with no-wash procedure required [38]. Meanwhile, Nile red as a good environment-sensitive fluorophore was also used for SNAP-tag-based protein labeling, although it can work only in the plasma membrane [39]. The great advantage of environment-sensitive fluorogenic probe was that this system avoids complicated modification of BG-substrate while the fluorogenic probe displayed significant turn-on effect and rapid labeling reaction rate.
2.3. In situ fluorophore generationHowever, there are some disadvantages for quencher release mechanism and environment-sensitive fluorophores. Most of the quencher-based fluorogenic probes required complex synthesis and the large molecule size often lead to low cell permeability and long protein labeling time. Environment-sensitive fluorophores normally displayed weak brightness and photostability which limited their applications in live-cell imaging. Consequently, the in situ generation of excellent fluorophores is an effective method to create better fluorogenic probes (Fig. 2c). Johnsson and co-workers made important contributions in advancing the techniques of in situ fluorophore formation. The methyl group at the 2-position of the phenyl ring from SiR-methyl was changed to a carboxyl group and SiR-carboxyl was created (Fig. 3c). They found that the reaction of SiR-carboxyl BG-derivative with SNAP-tag keep the fluorophore in its fluorescent zwitterionic form, while unreacted, aggregated or nonspecifically bound probe exists predominantly as the nonfluorescent spirolactone [40]. Meanwhile, the uncharged spirolactone increased the membrane permeability of the probe, which made it readily pass through the plasma membrane as well as internal membranes. The key feature for cellular applications is that fluorescence imaging of SiR-SNAP-tag protein labeling can be performed without removing excess fluorescent substrate through a washing process. In addition, its far-red emission and fluorogenicity make it ideally suited for applications in tissue samples and in vivo imaging [41]. Subsequently, Grimm et al. further improved the spectroscopic properties of the fluorophore by replacing of the N, N-dimethylamino substituents with azetidines in SiR, which increased quantum efficiencies and photostability [42]. The fluorogenic and spectroscopic properties of SiR-carboxyl make the dye attractive for applications beyond SNAP-tag labeling. This has been convincingly demonstrated by the attachment of SiRcarboxyl fluorophore to various targeting ligands (such as Halo-tag, CLIP-tag, antibody, and un-natural amino acids) for live-cell imaging of the cytoskeleton, various organelles, and nucleus as well as receptor proteins, etc. [43-46].
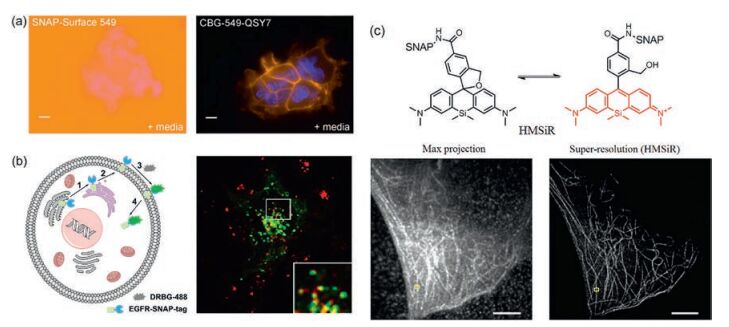
3. Applications of SNAP-tag fluorogenic probes 3.1. Real-time protein labeling in living cellsIn the cases discussed above, besides SiR-carboxyl, most of the switching properties of the designed probes are exploited after coupling of the probe to SNAP-tag. These fluorogenic substrates have been demonstrated for the utility in live-cell imaging of various organelles, nucleus and cell membrane without removing excess substrate by washing steps (Fig. 4a) [32, 34, 36]. Furthermore, based on the rapid labeling reaction rate, these fluorogenic substrates were versatile research tools to study protein dynamics behaviors in living systems. Additionally, if the substrate is membrane impermeable, the appearance of cell surface proteins at the plasma membrane in real time can be monitored. For example, using a non-fluorescent substrate of SNAP-tag, DRBG-488 (Fig. 3a), Urano et al. characterized the removal and insertion of epidermal growth factor receptor (EGFR) in real-time and shed light on the spatiotemporal dynamics of EGFR endocytosis during cell migration (Fig. 4b) [33]. Meanwhile, a cell-permeable probe, DRBGFL-DA, was also developed for the fluorogenic detection of cytosolic proteins, although the labeling reaction was carried out over a relatively long incubation time of 3 h. Therefore, fluorogenic substrates should be more suitable in site-specific animal studies, where excess substrate cannot be easily washed out.

|
Download:
|
| Fig. 4. Application of the SNAP-tag fluorogenic probes in live-cells imaging without a washout step. (a) Comparison of SNAP-Surface 549 and CBG-549-QSY7 substrates for labeling SNAPf-EGFR in living cells. (b) The process of a membrane trafficking study withDRBG-488: (1) SNAP-EGFR was synthesized in rough ER, (2) delivered to plasma membrane via Golgi apparatus, (3) DRBG-488 was labeled to SNAP on plasma membrane and fluorescence turn-on, (4) SNAP-EGFR was delivered to endosome/lysosome for degradation. (c) Chemical structure and reversible equilibrium of HMSIR. β-Tubulin-SNAP fusion protein was expressed in HeLa cells and the max projection image and SLM super-resolution image was obtained with HMSiR-BG labeling. | |
{kind=link}
3.2. Super-resolution fluorescence imaging
In the last decade, imaging techniques have been significantly developed from conventional fluorescence microscopy to superresolution microscopy breaking through the diffraction limit of light [47, 48]. The cellular structure could be observed at unprecedented resolution by super-resolution techniques, including single-molecule localization microscopy (SLM), stimulated emission depletion (STED) microscopy, and structured illumination microscopy (SIM). SLM approaches, such as photoactivated localization microscopy (PALM) [49], stochastic optical reconstruction microscopy (STORM) [50], ground-state depletion microscopy followed by individual molecular return (GSDIM) [51], and direct STORM (dSTORM) [52] enable the construction of super-resolution images by site-specific localization and repeated detection of individual fluorophores. On the other hand, the fluorophores should switch between a fluorescent and a nonfluorescent state, detectable and localizable at any given time [7]. So far, SNAP-tag-containing protein labeling technology has been a powerful tool to load fluorophores to site-specific localization in cells for single-molecule live-cell imaging. A key attribution of all SLM methods is the development of new synthetic probes with excellent photochemical or photophysical properties such as high brightness, good absorptivity, excellent photostability, and in particular, the ability of fluorescence on-off switch. Due to the excellent fluorescent properties, HMSiR has been recognized as a spontaneously blinking fluorophore for live-cell SLM [41]. Most HMSiR molecules exist in a non-fluorescent state at physiological pH and blink spontaneously with a suitable lifetime of the fluorescent open form for detection. Subsequently, HMSiR-BG derivative was synthesized for dSTORM imaging in living cells. HeLa cells expressing SNAP-tagged β-tubulin were observed after vital staining with HMSiR-BG. It demonstrated that the probes with HMSiR fluorophore can be used to label specific target proteins in living cells (Fig. 4c).
4. ConclusionsThe method of covalent labeling of proteins has created a powerful tool to link synthetic organic molecule to biomacromolecules in living cells. SNAP-tag has been widely used as an excellent protein labeling tag. By applying various strategies to switch fluorescence, fluorogenic probes for SNAP-tag have been successfully developed to study protein localization and protein dynamics with super resolution microscopy in living cells. This site-specific labeling system is particularly applicable in systems where sensitive detection is required, for example, the protein quantification and site-specific animal studies, or in highthroughput screening platforms where washout is undesirable or impossible. The probe design can be easily adjusted as required by means of chemical modification, and thus, we believe more and better probes will be created with these design strategies. We can expect that SNAP-tag probes with a rapid fluorogenic response to intracellular POI will expand the possibilities and potential for the biological applications to investigate diverse cellular events associated with proteins.
AcknowledgmentsWe thank financial supports from the National Natural Science Foundation of China (Nos. 21422606 and 21502189) and Dalian Cultivation Fund for Distinguished Young Scholars (Nos. 2014J11JH130 and 2015J12JH205).
| [1] |
D.M. Chudakov, M.V. Matz, S. Lukyanov, K.A. Lukyanov, Physiol. Rev. 90(2010) 1103-1163. DOI:10.1152/physrev.00038.2009 |
| [2] |
P. Dedecker, F.C. De Schryver, J. Hofkens, J. Am. Chem. Soc. 135(2013) 2387-2402. DOI:10.1021/ja309768d |
| [3] |
A. Miyawaki, Nat. Rev. Mol. Cell Biol. 12(2011) 656-668. DOI:10.1038/nrm3199 |
| [4] |
L.D. Lavis, R.T. Raines, ACS Chem. Biol. 9(2014) 855-866. DOI:10.1021/cb500078u |
| [5] |
C.L. Thomas, A.J. Maule, J. Gen. Virol. 81(2000) 1851-1855. DOI:10.1099/0022-1317-81-7-1851 |
| [6] |
I. R. Correa Jr., Curr. Opin. Chem. Biol. 20(2014) 36-45.
|
| [7] |
L. Xue, I.A. Karpenko, J. Hiblot, K. Johnsson, Nat. Chem. Biol. 11(2015) 917-923. DOI:10.1038/nchembio.1959 |
| [8] |
D. Li, L. Liu, W.H. Li, ACS Chem. Biol. 10(2015) 1054-1063. DOI:10.1021/cb5007536 |
| [9] |
G.V. Los, L.P. Encell, ACS Chem. Biol. 3(2008) 373-382. DOI:10.1021/cb800025k |
| [10] |
A. Keppler, S. Gendreizig, T. Gronemeyer, et al., Nat. Biotechnol. 21(2003) 86-89. DOI:10.1038/nbt765 |
| [11] |
D. Srikun, A.E. Albers, C.I. Nam, A.T. Iavarone, C.J. Chang, J. Am. Chem. Soc. 132(2010) 4455-4465. DOI:10.1021/ja100117u |
| [12] |
C. Jing, V.W. Cornish, ACS Chem. Biol. 8(2013) 1704-1712. DOI:10.1021/cb300657r |
| [13] |
Y. Hori, T. Norinobu, M. Sato, et al., J. Am. Chem. Soc. 135(2013) 12360-12365. DOI:10.1021/ja405745v |
| [14] |
S. Mizukami, S. Watanabe, Y. Akimoto, K. Kikuchi, J. Am. Chem. Soc. 134(2012) 1623-1629. DOI:10.1021/ja208290f |
| [15] |
Y. Hori, K. Nakaki, M. Sato, S. Mizukami, K. Kikuchi, Angew. Chem. Int. Ed. 51(2012) 5611-5614. DOI:10.1002/anie.201200867 |
| [16] |
S. Mizukami, S. Watanabe, Y. Hori, K. Kikuchi, J. Am. Chem. Soc. 131(2009) 5016-5017. DOI:10.1021/ja8082285 |
| [17] |
S. Mizukami, T. Yamamoto, A. Yoshimura, S. Watanabe, K. Kikuchi, Angew. Chem. Int. Ed. 50(2011) 8750-8752. DOI:10.1002/anie.201103775 |
| [18] |
T. Gronemeyer, G. Godin, K. Johnsson, Curr. Opin. Biotechnol. 16(2005) 453-458. DOI:10.1016/j.copbio.2005.06.001 |
| [19] |
B. Mollwitz, E. Brunk, S. Schmitt, et al., Biochemistry 51(2012) 986-994. DOI:10.1021/bi2016537 |
| [20] |
T. Gronemeyer, C. Chidley, A. Juillerat, C. Heinis, K. Johnsson, Protein Eng. Des. Sel. 19(2006) 309-316. DOI:10.1093/protein/gzl014 |
| [21] |
C. Chidley, H. Haruki, M.G. Pedersen, E. Muller, K. Johnsson, Nat. Chem. Biol. 7(2011) 375-383. DOI:10.1038/nchembio.557 |
| [22] |
J. Kohl, J. Ng, S. Cachero, et al., Proc. Natl. Acad. Sci. U. S. A. 111(2014) E3805-E3814. DOI:10.1073/pnas.1411087111 |
| [23] |
M.A. Brun, K.T. Tan, R. Griss, et al., J. Am. Chem. Soc. 134(2012) 7676-7678. DOI:10.1021/ja3002277 |
| [24] |
A. Gautier, E. Nakata, G. Lukinavicius, K.T. Tan, K. Johnsson, J. Am. Chem. Soc. 131(2009) 17954-17962. DOI:10.1021/ja907818q |
| [25] |
J.P. Wei, X.L. Chen, X.Y. Wang, et al., Chin. Chem. Lett. 28(2017) 1290-1299. DOI:10.1016/j.cclet.2017.01.007 |
| [26] |
Y. Hori, K. Kikuchi, Curr. Opin. Chem. Biol. 17(2013) 644-650. DOI:10.1016/j.cbpa.2013.05.015 |
| [27] |
S. Mizukami, Y. Hori, K. Kikuchi, Acc. Chem. Res. 47(2014) 247-256. DOI:10.1021/ar400135f |
| [28] |
X.M. Li, R.R. Zhao, Y. Yang, et al., Chin. Chem. Lett. 28(2017) 1258-1261. DOI:10.1016/j.cclet.2016.12.029 |
| [29] |
L.L. Zhang, H.K. Zhu, C.C. Zhao, X.F. Gu, Chin. Chem. Lett. 28(2017) 218-221. DOI:10.1016/j.cclet.2016.07.008 |
| [30] |
W. Feng, Q.L. Qiao, S. Leng, et al., Chin. Chem. Lett. 27(2016) 1554-1558. DOI:10.1016/j.cclet.2016.06.016 |
| [31] |
K. Stöhr, D. Siegberg, T. Ehrhard, et al., Anal. Chem. 82(2010) 8186-8193. DOI:10.1021/ac101521y |
| [32] |
C.J. Zhang, L. Li, G.Y.J. Chen, Q.H. Xu, S.Q. Yao, Org. Lett. 13(2011) 4160-4163. DOI:10.1021/ol201430x |
| [33] |
T. Komatsu, K. Johnsson, H. Okuno, et al., J. Am. Chem. Soc. 133(2011) 6745-6751. DOI:10.1021/ja200225m |
| [34] |
X. Sun, A. Zhang, B. Baker, et al., Chembiochem 12(2011) 2217-2226. DOI:10.1002/cbic.201100173 |
| [35] |
C. Reichardt, Chem. Rev. 94(1994) 2319-2358. DOI:10.1021/cr00032a005 |
| [36] |
T.K. Liu, P.Y. Hsieh, Y.D. Zhuang, et al., ACS Chem. Biol. 9(2014) 2359-2365. DOI:10.1021/cb500502n |
| [37] |
Y.S. Zeng, R.C. Gao, T.W. Wu, C. Cho, K.T. Tan, Bioconjug. Chem. 27(2016) 1872-1879. DOI:10.1021/acs.bioconjchem.6b00290 |
| [38] |
S. Leng, Q. Qiao, L. Miao, et al., Chem. Comm. 53(2017) 6448-6451. DOI:10.1039/C7CC01483J |
| [39] |
E. Prifti, L. Reymond, M. Umebayashi, et al., ACS Chem. Biol. 9(2014) 606-612. DOI:10.1021/cb400819c |
| [40] |
G. Lukinavicius, K. Umezawa, N. Olivier, et al., Nat. Chem. 5(2013) 132-139. DOI:10.1038/nchem.1546 |
| [41] |
G. Yang, F. de Castro Reis, M. Sundukova, et al., Nat. Methods 12(2015) 137-139. |
| [42] |
S.N. Uno, M. Kamiya, T. Yoshihara, et al., Nat. Chem. 6(2014) 681-689. |
| [43] |
R.S. Erdmann, H. Takakura, A.D. Thompson, et al., Angew. Chem. Int. Ed. 53(2014) 10242-10246. DOI:10.1002/anie.201403349 |
| [44] |
G. Lukinavičius, C. Blaukopf, E. Pershagen, et al., Nat. Commun. 6(2015) 8497-8503. DOI:10.1038/ncomms9497 |
| [45] |
E. Kim, K.S. Yang, R.J. Giedt, R. Weissleder, Chem. Commun. 50(2014) 4504-4507. DOI:10.1039/c4cc00144c |
| [46] |
G. Lukinavicius, L. Reymond, E. D'Este, al., Nat. Methods 11(2014) 731-733. DOI:10.1038/nmeth.2972 |
| [47] |
B. Huang, H. Babcock, X. Zhuang, Cell 143(2010) 1047-1058. DOI:10.1016/j.cell.2010.12.002 |
| [48] |
S.W. Hell, Science 316(2007) 1153-1158. DOI:10.1126/science.1137395 |
| [49] |
E. Betzig, G.H. Patterson, R. Sougrat, et al., Science 313(2006) 1642-1645. DOI:10.1126/science.1127344 |
| [50] |
M.J. Rust, M. Bates, X. Zhuang, Nat. Methods 3(2006) 793-796. DOI:10.1038/nmeth929 |
| [51] |
J. Folling, M. Bossi, H. Bock, et al., Nat. Methods 5(2008) 943-945. DOI:10.1038/nmeth.1257 |
| [52] |
M. Heilemann, S. van de Linde, M. Schüttpelz, et al., Angew. Chem. Int. Ed. 47(2008) 6172-6176. DOI:10.1002/anie.v47:33 |