 2017, Vol. 28
2017, Vol. 28 


 ,
Chandrani Natha,
Narayana Murthy Gantaa,
Gulberk Ucarb,
Barij Nayan Sinhaa,
Venkatesan Jayaprakasha
,
Chandrani Natha,
Narayana Murthy Gantaa,
Gulberk Ucarb,
Barij Nayan Sinhaa,
Venkatesan Jayaprakasha
b Department of Biochemistry, Faculty of Pharmacy, Hacettepe University, Sıhhiye 06100, Ankara, Turkey
Monoamine oxidase (MAO) inhibitors introduced into clinical practice during 1960's were abandoned due to adverse effects, Such as hepatotoxicity, and "cheese effect", which was characterized by hypertensive crisis [1, 2]. These adverse effects are due to non-selective inhibition of MAO-isoforms. Development of newer inhibitors that inhibits MAO selectively and reversibly is thus a rational approach in overcoming the adverse effects reported with earlier MAO-inhibitors. Aurones, 2-benzylidenebenzofuran-3-(2H)-ones, are a subclass of flavonoids, recently research group synthesized a series of aurones and evaluated there in vitro rMAOB (rat MAO) inhibitory activity in the micromolar range [3]. Earlier few researchers synthesized flavones, thioflavones, and flavanones and evaluated as potential inhibitors of monoamine oxidase isoforms (hMAO-A and -B, human MAO) [4]. Our group has also reported a series of flavones having inhibitory potential on hMAO isoforms [5]. The objective of the presented work is to evaluate the inhibitory activity of anologues aurones against MAO isoforms. The results were compared and molecular docking simulation studies were also performed. In the presented work evaluate their respective aurones are reported. Design strategy for designing MAO inhibitors is shown in Fig. 1.

|
Download:
|
| Fig. 1. Design strategy adopted for designing MAO inhibitors (1–20) | |
2. Results and discussion 2.1. Chemistry
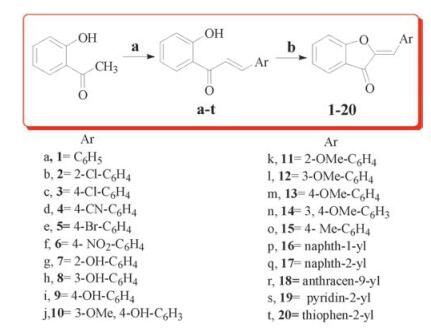
All the compounds were synthesized according to the reactions outlined in Scheme 1. Chalcones intermediates (a-t) were prepared through Claisen-Schmidt condensation of appropriate aromatic/heteroaromaticaldehydes with 2-hydroxyacetophenone in 60% aqueous ethanolic sodium hydroxide solution. The final products (1-20) were obtained by refluxing the chalcones (a-t), with mercuric acetate in pyridine at 110 ℃ and characterized by 1H NMR, 13C NMR and mass spectroscopy. 1H NMR of all the compounds, the olefinic proton (=CH-Ar) appeared as a singlet between δ 6.87-7.25, the hydroxyl proton of compounds 7-9 appeared as singlet between δ 10.20-12.37, the methoxy protons of compounds 10-14 appeared as singlet between δ 3.50-3.90, while methyl protons of compound 15 appeared as a singlet at δ 2.3. 13C NMRcharacterization has been carried out for group of compounds (6-8, 16-17 and 20) representing the enumerated library of twenty compounds (1-20). Compounds displayed carbonyl carbon (C-3) peak and (C-8) peak between δ 183.19-184-34 and δ 165.2-166.19 respectively. For compound 18, the carbonyl carbon (C-3) peak appeared at δ 169.66, while and (C-8) peak at δ 95.67. For compounds 7 and 8, the carbon having phenolic group exhibited the peak at δ 157.48 and δ 158.10, respectively. Mass spectra of all the compounds displayed M+ as molecular ion peak, except 2 (M +1)+.

|
Download:
|
| Scheme1. Reagents and conditions: (a) Ar-CHO, aq. NaOH (60%), EtOH, 48 h, r.t.; (b) Hg(OAc)2, pyridine, 110 ℃, 1 h | |
2.2. Biochemistry
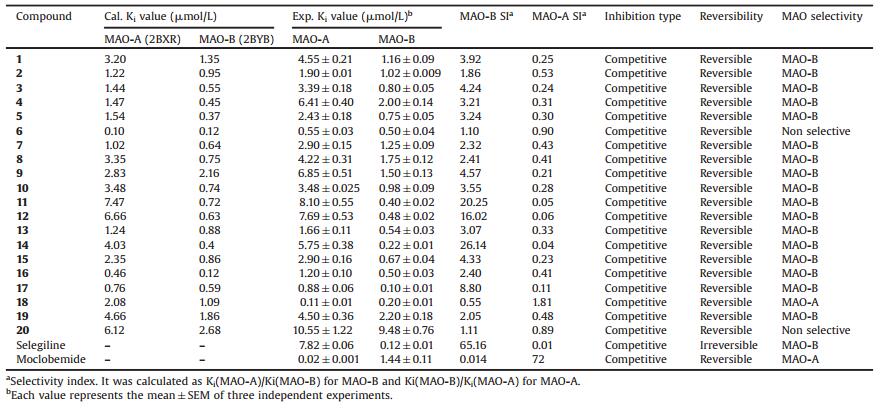
All the synthesized compounds 1-20 were screened for their hMAO inhibitory activity [6, 7] using recombinant human MAO isoforms. hMAO inhibitory activity was determined by measuring the production of H2O2 from p-tyramine, the common substrate for both hMAO-A and hMAO-B, using the Amplex®-Red MAO assay kit. The compounds and reference inhibitors did not react directly with the Amplex®-Red reagent indicating that they do not interfere with the measurements. The compounds were also confirmed, as they did not interact with resorufin by treating the maximum concentration of compounds with various concentrations of resorufin in order to detect if the fluorescence signal is the same with or without our compounds in the medium. No significant quenching of resorufin was observed. The experimental data concerning to the inhibition of hMAO-A and hMAO-B inhibition by the novel compounds 1-20 were presented in Table 1, together with their selectivity indices. The selectivity index was expressed as SI = Ki (MAO-A)/Ki (MAO-B) for selectivity towards hMAO-B. Protein was determined according to the method of Bradford method [8] using bovine serum albumin as the standard.
|
|
Table 1 Human monoamine oxidase inhibitory activity of compounds (1–20) |
{kind=link}
{kind=link}
Compounds, 1-5, 7-17 and 19 were found to inhibit hMAO-B selectively and reversibly, while, compound 18 having bulkier aldehyde portion (anthracen-9-yl) was found to be competitive, reversible and selective inhibitor of hMAO-A isoforms with Ki = 0.11 ±0.01 mmol/L, while compound 6 and 20 which carrying 4-NO2, and thiophen-2-yl were found to be nonselective towards the MAO-isoforms. The most potent hMAO-B inhibitor in this series was compound 17 (Ki = 0.10 ± 0.01 mmol/L), which was carrying naphth-2-yl group at aldehyde position. It was also found to have potency and selectivity almost equal to selegiline, a well-known selective MAO-B inhibitor (Ki = 0.12 ± 0.01 mmol/L), Compound 14 which was carrying 3, 4-diOMe functional group exhibited the best selectivity (SI = 26.14) towards hMAO-B. The activity profiles of this series of compounds were then compared with their flavone counterparts reported earlier by our group [5]. In spite of structural variation viz: (ⅰ) fused furan in place of pyran and (ⅱ) exocyclic double bond in place of endocyclic double bond, this series displayed similar activity profile as that of their flavone counterparts in potency as well as selectivity towards MAO isoforms.
A comprehensive SAR study revealed the following: (ⅰ) moving from phenyl (1) to 9-anthracenyl (18) substitution through 1-naphthyl (16)—potency increases but selectivity reversed from hMAO-B (1, 16)tohMAO-A(18).Interestingly, 2-naphthylderivative (17) was found to have potency equivalent to 18 with improved selectivity towards hMAO-B than 1. (ⅱ) Electron withdrawing functional groups at o-or p-positions did not exhibit significant improvement in either the potency or the selectivity towards hMAO-B for chloro (2 and 3), bromo (5) and cyano (4) substitutions in comparison with compound 1. The only exception is with compound having nitro group at p-position (6), which displayed improved potency with loss of selectivity in comparison with compound 1. (ⅲ) Electronpumping functional groups: (a) Hydroxyl group at o-, m-and p-(7-9) did not displayed significant improvementineitherpotencyofselectivity.(b)Replacinghydroxyl group with methoxy (11-13) improved the potency as well as selectivity towards hMAO-B to 3-4 fold & 7-9 fold, respectively for o-and m-substituted derivatives (11 and 12). p-Substituted derivative (13) displayed improved potency with no improvement in selectivity towards hMO-B. A similar effect is observed with the methyl substitution at p-position (15) too. (c), while 13 was displaying increase in potency, the selectivity was found to be improved by the additional methoxy group at m-position (14). Almost 18-fold improvement was observedand this compound was found to have the best selectivity within the series. (ⅳ) Replacing the phenyl ring of 1 with 2-pyridyl (19) or 2-thiophenyl (20) was found to decrease the potency as well as selectivity. Compound 19 was found to be non-selective similar to compound 6. This clearly establishes that deactivated ring (p-nitrophenyl of 6 and 2-pyridyl of 19) makes the molecule to lose its selectivity towards isoforms in comparison with compound 1.
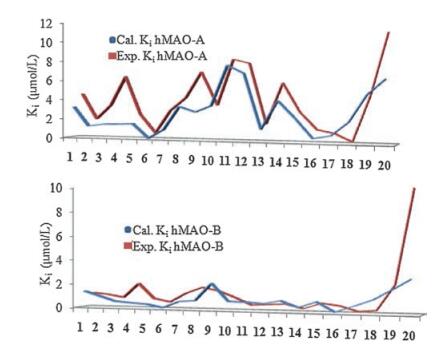
2.3. Molecular docking simulationMolecular docking simulation was carried to understand the effect of structural features that determines the potency and selectivityat molecular level. The approach has been adopted by other groups as well as our group in our earlier communications [5, 9-13]. The molecular docking simulation was done using AutoDock-4.2 by adopting the protocol reported earlier by our group. Estimated Ki valuesobtained were in good agreement with the experimental Ki values (Fig. 2).

|
Download:
|
| Fig. 2. Experimental and calculated Ki values corresponding to the inhibition of hMAO-isoforms with aurone derivatives | |
{kind=link}
2.3.1. Orientation of compounds (1-20) in active site of hMAO-B
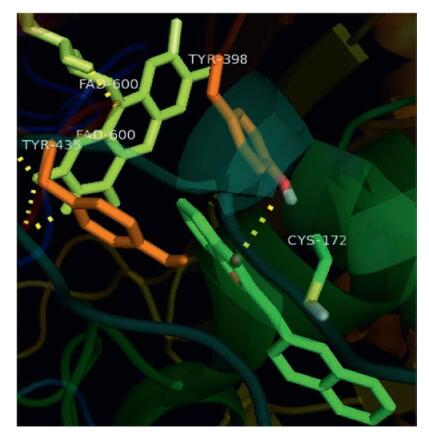
Active site of hMAO-B is hydrophobic in nature and is designated to have an aromatic cage lined by Flavin adenine dinucleotide (FAD), TYR435, and TYR398. The narrow space (hydrophobic tunnel) between aromatic cage and the active site entrance is lined with hydrophobic amino acid residues (LEU171, TYR326, PHE168, ILE198, and ILE199). Volume is relatively small when compared with the active site of hMAO. All the synthesized compounds (except 16, 18) oriented in a similar fashion as that of the potent hMAO-B inhibitor 17, the benzofuran-3(2H)-one portion oriented towards aromatic cage and the benzylidene portion was placed in the hydrophobic tunnel. The orientation is quiet similar to the one reported by Morales-Camilo et al. [3]. Compounds 16 and 18 exhibited reversed orientation with reference to compound 17. i.e., now the benzofuran-3(2H)-one portion was placed in the tunnel orienting the benzylidene portion in the aromatic cage. This unusual reversal due to ortho-fusion at 2, 3-position may be the reason why these molecules are less selective towards hMAO-B/ selective towards hMAO-A. The interactions of the molecules are purely hydrophobic and they exhibited π-π stacking interaction with components of aromatic cage. Fig. 3 displays the interaction of compound 17 with TYR435 (12.03 Å) and FAD (30.35 Å).

|
Download:
|
| Fig. 3. Interaction of compound 17 with hMAO-B (PDB: 2BYB) active site | |
{kind=link}
2.3.2. Orientation of compounds (1-20) in active site of hMAO-A
Active site of hMAO-A is hydrophobic in nature as that of hMAO-B. Volume of the pocket is larger with three sub-pockets namely (ⅰ) pocket 1: aromatic cage lined by FAD, Tyr407 & Tyr444; (ⅱ) pocket 2: lined by Ile180, Ile335, Leu337, Met350 & Phe352 and (ⅲ) pocket 3: lined by Gly74, Arg206, Ile207, Phe208, Glu216 and Trp441. This could able to accommodate larger molecules. Compounds 1, 2, 7-9, 16, 19 and 20, the benzylideneportion orient towards pocket 1, while the benzofuran-3(2H)-one portion orient towards pocket 3 and left the pocket 2 unoccupied. In case of compounds 3, 10-12 and 14, the benzofuran-3(2H)-one portion orient towards pocket 1 placing the benzylidene portion at the center leaving pocket 2 and pocket 3 unoccupied. While in compounds 4-6, 13 and 15, the benzylidene portion was found to completely fill the pocket 3 which keeps the benzofuran-3(2H)-one portion to accommodate in pocket 1 leaving pocket 2 unoccupied. Compound 17 places the entire molecule in the axis that connects aromatic cage with the entrance, leaving pockets 2 and 3 unoccupied. Incomplete occupation of the sub-pockets seems to be the reason behind the poor interaction of ligand with the protein leading to reduced potency and selectivity for the compounds in this series. Surprisingly the most potent hMAO-A inhibitor of this series, compound 18 that did not occupy any pockets but it resided at the entrance lined by THR201, VAL210, TYR121, TRP128, PHE208, TYR124, GLY110 and Phe173. The bulkier anthracene ring found to close the entrance cavity displaying hydrophobic interaction with the hydrophobic residues at the entrance and projecting the rest of the molecules into the entrance cavity. This resembles the cork closing the wine-bottle.
3. ConclusionA series of twenty 2-(arylmethylidene)-2, 3-dihydro-1-benzofuran-3-one derivatives were synthesized and screened for their potential to inhibit hMAO isoforms selectively. With the limited variations at single site, benzylidene portion the activity profile was recorded and a concise SAR was derived. In summary, weak to moderate electron pumping as well as electron withdrawing groups favor selectivity towards hMAO-B, while strong deactivators make them non-selective towards isoforms. Mono-substitution of methoxy group at o-and m-position increases potency while bi-substitution lead to the improved potency as well as selectivity. The same may be extended to flavones reported in our earlier communication [5].
4. Experimental 4.1. Materials and methodsChemicals and solvents were of reagent grade and purchased from Sigma-Aldrich. Melting points were determined using OPTIMELT (SRS, Sweeden) an automated melting point system and are uncorrected melting point. 1H NMR spectra were recorded on Varian 400 MHz and ECX-500 (JEOL) 300 MHz, DMSO-d6 and CDCl3 as a solvent. In 1H NMR spectra the coupling constants (J) are expressed in hertz (Hz). Chemical shifts (δ) of NMR are reported in parts per million (ppm) units relative to the solvent. 13C NMR 100 MHz, in DMSO-d6 and CDCl3 as a solvent. Mass spectra were recorded in WATERS-Q-Tof Premier-HAB213 and ESI-MS technique. hMAO-A and hMAO-B (both recombinant, expressed in baculovirus-infected BTI insect cells), Selegiline, Moclobemide, Resorufin, DMSO, and other chemicals were purchased from Sigma-Aldrich (Munich, Germany). The Amplex®-Red MAO assay kit containing benzylamine, p-tyramine, Clorgyline, Pargyline, and horseradish peroxidase was purchased from Cell Technology Inc., Mountain View, CA, USA. Molecular modelling studies were carried using AutoDock-4.2 on RHEL-5.0 Operating system installed on Dell Precision workstation with Intel core-2 quad processor and 8 GB RAM.
4.2. Chemistry 4.2.1. General procedure for synthesis of Chalcones (a-t)To a solution of 2-hydroxy acetophenone (0.01 mol/L) and appropriate benzaldehyde (0.01 mol/L) in ethanol (10 mL), was added 60% aq. sodium hydroxide solution at < 10 ℃ with continuous stirring for a period of 30-45 min. The reaction mixture was then left at room temperature for about 48 h with occasional shaking. After 48 h, the reaction mixture was poured into ice-cold water, and was then adjusted to pH 2 using 6 mol/L HCl. The yellow precipitate obtained was filtered, washed with water and dried. The obtained crude was purified by column chromatography and then recrystallized from ethanol to give yellow product [14-16].
4.2.2. General procedure for synthesis of compounds (1-20)To a solution of mercuric acetate (0.1 mol/L) in pyridine (10 mL) was added chalcone (0.1 mol/L) at room temperature and the mixture was refluxed at 110 ℃ for 1 h. After completion, the reaction mixture was poured into ice cold water and acidified with hydrochloric acid (10% aqueous solution). The precipitated solid was extracted with DCM, the extracts were dried on sodium sulfate and the solvent was evaporated to give a solid which was further purified by column chromatography [17].
4.3. Biochemistry 4.3.1. Determination of hMAO-A and -B activitiesThe activities of hMAO-A and hMAO-B were determined using p-tyramine as common substrate and calculated as 177.00 -9.55 pmol/mg/min (n = 3) and 140.90 ±11.70 pmol/mg/min (n = 3), respectively. The interactions of the synthesized compounds with hMAO isoforms were determined by a fluorimetric method described and modified previously activity [6, 7]. The production of H2O2 catalyzed by MAO isoforms was detected using Amplex®-Red reagent, a non-fluorescent, highly sensitive, and stable probe that reacts with H2O2 in the presence of horseradish peroxidase to produce the fluorescent product resorufin. The reaction was started by adding (final concentrations) 200 mmol/L Amplex Red reagent, 1 U/mL horseradish peroxidase, and p-tyramine (concentration range 0.1-1 mmol/L). Control experiments were carried out by replacing the synthesized compound and reference inhibitors. The possible capacity ofcompounds to modify the fluorescence generated in the reaction mixture due to nonenzymatic inhibition was determined by adding these compounds to solutions containing only the Amplex Red reagent in a sodium phosphate buffer.
4.3.2. Kinetic experimentsSynthesized compounds were dissolved in DMSO, with a maximum concentration of 1%, and used in the concentration range of 1-100 mmol/L. Kinetic data for interaction of the enzyme isoforms with the compounds were determined using the Microsoft Excel package program. The specificity index was expressed as SI = Ki (MAO-A)/Ki (MAO-B). The protein was determined according to the Bradford method [8], in which bovine serum albumin was used as standard.
4.3.3. Reversibility experimentsReversibility of the MAO inhibition with synthesized compounds were evaluated by a centrifugation-ultrafiltration method [18]. In brief, adequate amounts of the recombinant hMAO-A or B were incubated together with a single concentration of the synthesized compounds or the reference inhibitors in a sodium phosphate buffer (0.05 mol/L, pH 7.4) for 1 h at 37 ℃. After this incubationperiod, an aliquot was stored at 4 ℃ and used for themeasurement of MAO-A and -B activity. The remaining incubatedsample was placed in an Ultrafree-0.5 centrifugal tube with a 30 kDa biomaxmembrane and centrifuged at 9000 × g for 20 min at 4 ℃. The enzyme retained in the 30 kDa membrane was resuspended in a sodium phosphate buffer at 4 ℃ and centrifuged again two successive times. After the third centrifugation, the enzymeretained in the membrane was resuspended in sodium phosphate buffer (300 mL) and an aliquot of this suspension was used forMAO-A and-B activity determination. Control experiments were performed simultaneously (to define 100% MAO activity) byreplacing the testdrugs with appropriate dilutions of the vehicles. The corresponding values of percent (%) MAO isoform inhibitionwas separately calculated for samples with and without repeatedwashing.
4.4. Molecular docking simulationIn order to understand the interaction at molecular level, compounds (1-20) were docked with X-ray crystal structure of hMAO-A (PDB: 2BXR) and hMAO-B (PDB: 2BYB) using AutoDock 4.2 following the protocol reported in our earlier communications [5, 9-13]. For docking, grid parameter file (.gpf) and docking parameter files (.dpf) were written using MGLTools-1.5.6. Receptor grids were generated using 60 × 60 × 60 grid points in xyz with grid spacing of 0.375 Å. Grid box was centered on N5 atom of FAD. Map types were generated using autogrid-4.2. Docking was carried out with following parameters: number of runs: 50, population size: 150, number of evaluations: 2, 500, 000 and number of generations: 27, 000, using autodock 4.2. Analysis of docking results was done using MGLTools-1.5.6. Top scoring molecule in the largest cluster was analyzed for its interaction with the protein.
AcknowledgmentsFirst author acknowledges to Birla Institute of Technology for providing financial support as a prestigious Institute fellowship, also acknowledge to Central Instrumentation Facility and NMR Facility at BIT, Mesra for Spectral Characterization of the compounds.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2017.02.009
| [1] | D. Robert Knapp. Cheese and monoamine oxidase inhibitorsHeadache. J. Head Face Pain 3 (1964) 157–158. DOI:10.1111/hed.1964.3.issue-4 |
| [2] | B. Blackwell. Hypertensive crisis due to monoamine-oxidase inhibitors. Lancet 282 (1963) 849–851. DOI:10.1016/S0140-6736(63)92743-0 |
| [3] | N. Morales-Camilo, C.O. Salas, C. Sanhueza, et al., Synthesis, biological evaluation, and molecular simulation of chalcones and aurones as selective MAO-B inhibitors. Chem. Biol. Drug Des. 85 (2015) 685–695. DOI:10.1111/cbdd.2015.85.issue-6 |
| [4] | F. Chimenti, R. Fioravanti, A. Bolasco, et al., A new series of flavones, thioflavones, and flavanones as selective monoamine oxidase-B inhibitors. Bioorg. Med. Chem. 18 (2010) 1273–1279. DOI:10.1016/j.bmc.2009.12.029 |
| [5] | V.N. Badavath, S. Ciftci-Yabanoglu, S. Bhakat, et al., Monoamineoxidaseinhibitory activity of 2-aryl-4H-chromen-4-ones. Bioorg. Chem. 58 (2015) 72–80. DOI:10.1016/j.bioorg.2014.11.008 |
| [6] | F. Chimenti, E. Maccioni, D. Secci, et al., Synthesis, stereochemical identification, and selective inhibitory activity against human monoamine oxidase-B of 2-methylcyclohexylidene-(4-arylthiazol-2-yl) hydrazones. J. Med. Chem. 51 (2008) 4874–4880. DOI:10.1021/jm800132g |
| [7] | M. Yanez, N. Fraiz, E. Cano, F. Orallo. Inhibitory effects of cis-and transresveratrol on noradrenaline and 5-hydroxytryptamine uptake and on monoamine oxidase activity. Biochem. Biophys. Res. Commun. 344 (2006) 688–695. DOI:10.1016/j.bbrc.2006.03.190 |
| [8] | M. Marion Bradford. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 (1976) 248–254. DOI:10.1016/0003-2697(76)90527-3 |
| [9] | V.N. Badavath, S. Ciftci-Yabanoglu, S.S. Jadav, M. Jagrat, B.N. Sinha, G. Ucar, V. Jayaprakash. Monoamine oxidase inhibitory activity of 3, 5-biaryl-4, 5-dihydro-1H-pyrazole-1-carboxylate derivatives. Eur. J. Med. Chem. 69 (2013) 762–767. DOI:10.1016/j.ejmech.2013.09.010 |
| [10] | V.N. Badavath, B. Ipek, G. Ucar, B.N. Sinha, V. Jayaprakash. Monoamine oxidase inhibitory activity of novel pyrazoline analogues: curcumin based design and synthesis. ACS Med. Chem. Lett. 7 (2016) 56–61. DOI:10.1021/acsmedchemlett.5b00326 |
| [11] | V.N. Badavath, G. Ucar, B.N. Sinha, S.K. Mondal, V. Jayaprakash. Monoamine oxidase inhibitory activity of novel pyrazoline analogues: curcumin based design and synthesis-Ⅱ. Chem. Sel. 1 (2016) 1–7. |
| [12] | V.N. Badavath, B. Ipek, G. Ucar, S.K. Mondal, B.N. Sinha, V. Jayaprakash. MAO inhibitory activity of ferulic acid amides: curcumin based design and synthesis. Arch. Pharm. 348 (2015) 1–11. DOI:10.1002/ardp.v348.1 |
| [13] | V.N. Badavath, B.N. Sinha, V. Jayaprakash. Design, in-silico docking and predictive ADME properties of novel pyrazoline derivatives with selective human MAO inhibitory activity. Int. J. Pharm. Pharm. Sci. 7 (2015) 277–282. |
| [14] | V.N. Badavath, S.S. Jadav, B. Pastorino, et al., Synthesis and antiviral activity of 2-phenyl-4H-chromen-4-one derivatives against Chikungunya virus. Lett. Drug Des. Discov. 13 (2016) 1–6. |
| [15] | T. Narender, K. Venkateswarlu, V.N. Badavath, S. Sarkar. A new chemical access for 3'-acetyl-4'-hydroxychalcones using BF3-Et2O via a regioselective Claisen-Schmidt condensation and its application in the synthesis of chalcone hybrids. Tetrahedron Lett. 52 (2011) 5794–5798. DOI:10.1016/j.tetlet.2011.08.120 |
| [16] | S.S. Jadav, S. Kaptein, T. Ajay kumar, et al., Design, synthesis, optimization and antiviral activity of a class of hybrid dengue virus E protein inhibitors. Bioorg. Med. Chem. Lett. 25 (2015) 1747–1752. DOI:10.1016/j.bmcl.2015.02.059 |
| [17] | S. Venkateswarlu, G.K. Panchagnula, A.L. Gottumukkala, G.V. Subbaraju. Synthesis, structural revision, and biological activities of 4'-chloroaurone a metabolite of marine brown alga Spatoglossumvariabile. Tetrahedron 63 (2007) 6909–6914. DOI:10.1016/j.tet.2007.04.048 |
| [18] | F. Chimenti, S. Carradori, D. Secci, et al., Synthesis and inhibitory activity against human monoamine oxidase of N1-thiocarbamoyl-3, 5-di(hetero)aryl-4, 5-dihydro-(1H)-pyrazole derivatives. Eur. J. Med. Chem. 45 (2010) 800–804. DOI:10.1016/j.ejmech.2009.11.003 |