 2017, Vol. 28
2017, Vol. 28 



b Department of Chemistry, International College of Zhengzhou University, Zhengzhou University, Zhengzhou 450001, China;
c Department of Chemistry, College of Chemistry and Chemical Engineering, The Key Laboratory for Chemical Biology of Fujian Province, Xiamen University, Xiamen 361005, China
Cancer has become an important public health concern and a significant cause of death in the human population [1]. Cancer incidence and mortality have been increasing in China, making cancer the leading cause of death since 2010 and a major public health problem in the country [2]. Lung cancer was the leading cause of death in China followed by liver cancer, stomach cancer, esophageal cancer and colorectal cancer [3]. Despite many efforts to fight against cancer, the successful treatment of certain tumor types continues to be a challenge owing to their aggressiveness, the mechanisms of malignant cell metastasis, chemoresistance, and the lack of selectivity of some drugs [4]. Therefore, the development of novel anticancer agents with high efficacy and minimal side effects by synthesizing small and simple molecules is indispensable.
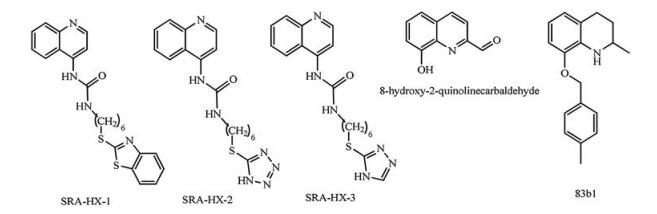
N-Heterocyclic compounds are very crucial in drug design [5-7]. Compounds containing quinoline rings are extensively found in several classes of natural and synthetic biologically active compounds [8-10]. Quinoline-bearing structures are well-known due to their broad biological activities [11], such as anticancer [12], antifungal [13], antibacterial [14], antitubercular [15] and antimalarial [16] that have been used in traditional medicine as a remedy. Recently, quinoline-based azolyalkylquinolines bearing different azole groups, such as benzothiazole (SRA-HX-1), tetrazole (SRAHX-2), and 1, 2, 4-triazole (SRAHX-3), have been synthesized and reported as potent antitumor agents for breast cancer cells in vitro [17]. 8-Hydroxyquinoline derivative (8-hydroxy-2-quinolinecarbaldehyde) has been reported to possess strong antitumor activities against human cancer cell lines and hepatocellular carcinoma Hep3B [18]. Moreover, a novel quinoline derivative 83b1 (8-(4-(trifluoromethyl)benzyloxy)-1, 2, 3, 4-tetrahydro-2-methylquin-oline, has shown to inhibit cancer growth in human esophageal squamous cell carcinoma [19]. The structure of the quinoline derivative is depicted in Fig. 1.

|
Download:
|
| Fig. 1. Quinoline derivatives. | |
α-Aminophosphonic acids and their corresponding α-amino phosphonates have received much attention in organic and medicinal chemistry because of their pharmacological properties and clinical applications [20-22]. Some of them have been used as potent enzyme inhibitors, antimicrobial, antitumor, antioxidant and antiviral agents [23-28]. It was reported that introduction of aminophosphonate group to pharmacy core is able to improve the antitumor activity and many aminophosphonate derivatives have demonstrated potent inhibitory activities against human tumors [29, 30].
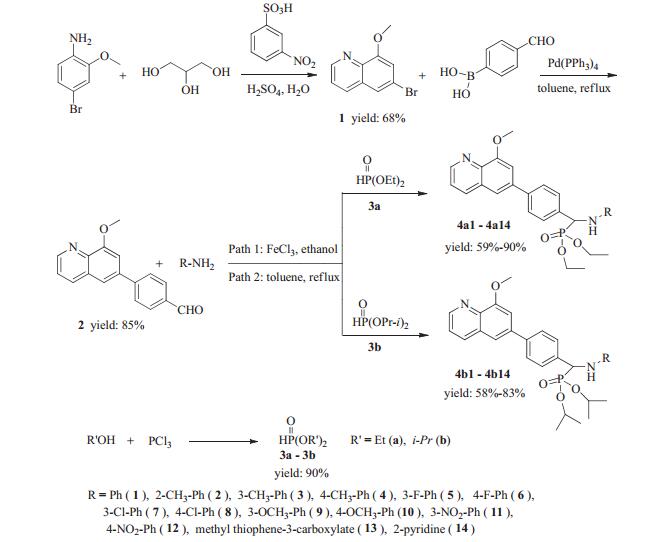
Existing thieno[2, 3-d]pyrimidine in one α-amino phosphonate molecule can exhibit valuable anti-tumor activity [31]. These special scaffold compounds strongly provoked our interest to continuously explore this kind of compounds. Based on this strategy, we lead to the proposal to incorporate quinoline scaffold into the α-aminophosphonate. Then two series of α-aminophosphonate derivatives containing a quinoline moiety were synthesized (Scheme 1). The in vitro cytotoxic activities of target compounds were tested against esophageal cancer (Eca109) and hepatocellular carcinoma (Huh7) by MTT method. This represents the first report about the synthesis and in vitro antitumor activity evaluation of α-aminophosphonate derivatives containing a quinoline moiety.

|
Download:
|
| Scheme1. The synthetic route for target compounds. | |
2. Results and discussion
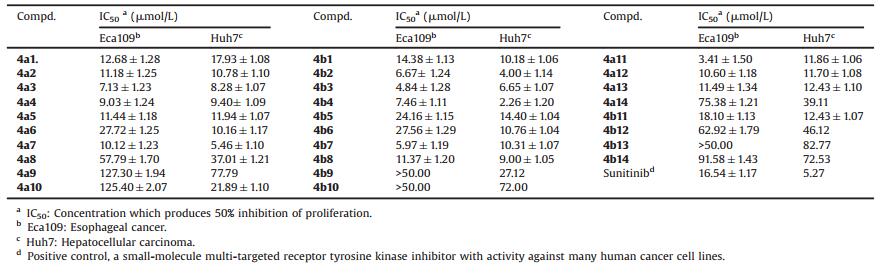
As shown in Table 1, the results of preliminary bioassay reveal that most of target compounds exhibited moderate to strong antitumor activity against human cancer cell lines Eca109 and Huh7 cells. Some of them demonstrated more potent antitumor activities than the reference drug. Sunitinib (SU) is a multitargeted tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Recently, Sunitinib has been used to treat solid cancers, such as renal cell carcinoma, gastrointestinal stromal tumors, neuroendocrine tumors in several Phase Ⅱ/Ⅲ trials, lung cancer, pancreatic cancer, chondrosarcoma, esophageal cancer, bladder cancer, glioma, aggressive fbromatosis, and also showed potential efficacy in progression-free survival and overall survival [32]. Furthermore, research shows that Sunitinib has same antitumor activity in hepatocellular carcinoma [33]. So Sunitinib was selected as the positive control.
|
|
Table 1 The cytotoxic activity of target compounds against human cancer cell lines. |
{kind=link}
{kind=link}
In Eca109 assay, compounds 4a1, 4a2, 4a5, 4a7, 4a12, 4a13, 4b1 and 4b8 exhibited preferable cytotoxic activity than the Sunitinib (IC50 = 16.54 μmol/L), implying favorable inhibition activities of these compounds on Eca109 cell. Besides, compounds 4a3, 4a4, 4b2, 4b3, 4b4 and 4b7 were approximately 2-3 times potent than Sunitinib. Interestingly, compound 4a11, the most promising compound, showed significant inhibitory preference to Eca109 with IC50 value of 3.41 μmol/L, about 5 times more potent than Sunitinib. In Huh7 assay, compounds 4a3, 4a4, 4a7, 4b2, 4b3, 4b4 and 4b8 showed potent antitumor activity. Especially, compounds 4b2 and 4b4 (IC50 = 2.26 μmol/L-4.00 μmol/L) showed more potent activity against Huh7 than Sunitinib (IC50 = 5.27 μmol/L). Among all the target compounds, it is worth noting that compounds 4b2 and 4b4 which possess methyl-substituted aniline group were found to be more active than Sunitinib against both of two cancer cell lines, with IC50 in the range of 2.26 μmol/L-7.46 μmol/L.
The antitumor data also indicate that the substituents of phosphonate have no apparent influence on antitumor activity to Eca109 and Huh7 cells. For example, when R was Ph and R' was Et, compound 4a2 exhibited good antitumor activities against Eca109 and Huh7 cells, with IC50 values of 11.18, 10.78 μmol/L, respectively. The inhibitory activity of compound 4b2, where the phosphonate was substituted by i-Pr, was litter higher than that of 4a2 against Eca109 and Huh7 with IC50 values of 6.67, 4.00 μmol/L, respectively. The same trend was observed for compounds 4a3/4b3, 4a4/4b4 and 4a8/4b8 against Eca109 and Huh7 cells. While compound 4a5 with 3-F (R) and Et (R') showed antitumor activity against Huh7 with IC50 values of 11.94 μmol/L. Compound 4b5 with 3-F (R) and i-Pr (R') had a lower activity (IC50 = 14.4 μmol/L) than that of 4a5. This also applied to compounds 4a11/4b11, 4a12/4b12 against Eca 109 and Huh7 cells.
Different substituents on the phenyl ring had a great influence in antitumor activity. Compound 4a1 and 4b1 with unsubstituted aryl ring exhibited good antitumor activities against Eca109 and Huh7 cells, with IC50 values of 12.68, 17.93 and 14.38, 10.18 μmol/L, respectively.Compounds 4a2-4a4 and 4b2-4b4 containing methyl-substituted aniline group showed more potent antitumor activities against the tested cancer cell lines with IC50 in the range of 2.26-11.18 μmol/L. Compounds 4a7, 4b7 and 4b8 with 3-chlorophenyl and 4-chlorophenyl moiety showed enhanced inhibitory activity against Eca109 andHuh7 cells, as well as compounds 4a11, 4b11 and 4a12 with 3-nitrophenyl and 4-nitrophenyl moiety. Besides, metasubstituted derivatives showed more potent activity than parasubstituted derivatives (4a7 vs. 4a8, 4a11 vs. 4a12, 4b11 vs. 4b12). These results indicated that methyl-, chloro-and nitro-substituent at thephenyl ring is favorable for activity.However, compounds 4a6, 4b5 and 4b6 with 3-fluoro phenyl and 4-fluoro phenyl moiety had less cytotoxic activity against Eca109 and Huh7 cells. The antitumor activities of compounds 4a9, 4a10, 4b9 and 4b10 with 3-methoxylphenyl and 4-methoxylphenyl moiety were decreased dramatically. These results indicated that methoxyl at the phenyl ring are unfavorable. When aromatic amine group was substituted by benzylamine or 2-aminopyridine, the target compounds 4a13, 4a14, 4b13 and 4b14 had less or inconspicuous cytotoxic activity.The bioactive assay implied that the substitutions on the phenyl ring significantly affect the antitumor activity of the target compounds.
3. ConclusionIn summary, two series of α-aminophosphonate derivatives containing a quinoline moiety were synthesized efficiently and their cytotoxic activities against two human cancer cell lines including Eca109 and Huh7 were evaluated for the first time. Moreover, based on experimental results, the structure-activity relationship was analyzed. Biological assays revealed that the substitutions on the phenyl ring influenced the antitumor activity remarkably, while the substitutes of phosphonate had no apparent influence on the antitumor activity. Among them, compound 4a11 showed excellent inhibitory activity to Eca109, approximately 5-fold more active than Sunitinib. Additionally, compounds 4b2 and 4b4 containing methyl-substituted aniline group were the most efficient against both of the tested cancer cell lines. All the above results demonstrated that these compounds could be promising lead compounds of novel antitumor drugs. Further studies will focus on structural optimization and precise mechanism of action of these compounds.
4. ExperimentalCommercial reagents and solvents were used as received without further purification unless otherwise specified. Toluene was freshly distilled over sodium with the use of diphenyl ketone as an indicator under nitrogen. High resolution mass spectra (HRMS) were obtained with a Q-Tof mass spectrometer using the ESI technique. NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer. 1H and 13C chemical shifts were quoted in DMSO-d6 with tetramethylsilane (TMS) as the internal standard, and 31P chemical shifts were acquired in DMSO-d6 with H3PO4 as the internal standard. Column chromatography was performed on silica gel 200-300 mesh.
The target compounds 4a1-4a14 and 4b1-4b14 were prepared as shown in Scheme 1. Compound 1 was synthesized according to well-established literature procedures [34]. Commercially available 4-bromo-2-methoxyaniline treated with glycerin in the presence of 3-nitrobenzenesulfonic acid with concentrated sulfuric acid to afford 6-bromo-8-methoxy-quinoline (1). Compound 1 then underwent Suzuki cross-coupling reaction with 4-formylphenylboronic acid in toluene, using Pd(PPh3)4 as a catalyst to give intermediate 2 with excellent yield. Various methods for the synthesis of α-aminophosphonates were reported. To our best knowledge, one pot Mannich-type process of carbonyl compounds, amines, and dialkyl phosphonate in the presence of a Lewis acid remains the most efficient, simple, general and high yielding method. Therefore, the reaction of intermediate 2 with diethyl or diisopropyl phosphonate and various substituted amines in the presence of FeCl3 in absolute ethanol generated target compounds (4a1-4a10 and 4b1-4b10) with good yields (Pathway 1). Unfortunately, the scope of this reaction was limited and the target compounds (4a11-4a14 and 4b11-4b14) were obtained by the reaction of intermediate 2, various amine with dialkyl phosphonate in anhydrous toluene by one pot synthesis without any catalyst (Pathway 2). All the newly synthesized compounds were characterized by 1H NMR, 13C NMR, 31P NMR, IR and high resolution mass spectrometry, respectively (see Supporting information for structure characterization).
For preliminary screening of antitumor candidates, the cytotoxic activities of target compounds were evaluated against two human cancer cell lines Eca109 and Huh7 using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay. The cancer cell lines seeded into the 96-well plate (100mL each well) were incubated at 37 ℃ in a 5% CO2 incubator. After 24 h, the target compounds at different concentrations were added to the culture medium and the cell cultures were continued for 72 h. The cultured cells were mixed with 10 μL 5 mg/mL of MTT solution and incubated for 4 h at 37 ℃. The formazan crystals were dissolved in 100 mL DMSO each well, and the absorbency at 570 nm and 630 nm (for the reference wavelength) was measured with microplate reader. Each experiment was performed at least three times. The results expressed as IC50 (inhibitory concentration 50%) were the averages of three determinations and calculated by using the GraphPad Prism 6.0 software. The results were illustrated in Table 1 with Sunitinib as the positive control.
AcknowledgmentWe gratefully acknowledge financial support of this work by the National Natural Science Foundation of China (Nos. 21105091, 21172201).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.02.012.
| [1] | A. Jemal, F. Bray, M.M. Center, et al., Global cancer statistics, CA Cancer. J. Clin. 61 (2011) 69–90. DOI:10.3322/caac.v61:2 |
| [2] | China National Statistical Bureau. China Statistical Yearbook-2010, Beijing, China: China Statistics Press, 2010 . |
| [3] | W.Q. Chen, R.S. Zheng, H.M. Zeng, et al., Annual report on status of cancer in China, 2011. Chin. J. Cancer Res. 27 (2015) 2–12. |
| [4] | G. Colombano, C. Travelli, U. Galli, et al., A novel potent nicotinamide phosphoribosyltransferase inhibitor synthesized via click chemistry. J. Med. Chem. 53 (2010) 616–623. DOI:10.1021/jm9010669 |
| [5] | V. Spanò, A. Montalbano, A. Carbone, et al., Synthesis of a new class of pyrrolo [3, 4-h]quinazolines with antimitotic activity. Eur. J. Med. Chem. 74 (2014) 340–357. DOI:10.1016/j.ejmech.2013.10.014 |
| [6] | R. Dua, S. Shrivastava, S.K. Sonwane, S.K. Srivastava. Pharmacological significance of synthetic heterocycles scaffold: a review. Adv. Biol. Res. 5 (2011) 120–144. |
| [7] | P. Diana, A. Carbone, P. Barraja, et al., Synthesis and antitumor activity of 3-(2-phenyl-13-thiazol-4-yl)-1 H-indoles and 3-(2-phenyl-1, 3-thiazol-4-yl)-1 H-7-azaindoles. ChemMedChem 6 (2011) 1300–1309. DOI:10.1002/cmdc.v6.7 |
| [8] | K.D. Thomas, A.V. Adhikari, N.S. Shetty. Design synthesis and antimicrobial activities of some new quinoline derivatives carrying 1, 2, 3-triazole moiety. Eur. J. Med. Chem. 45 (2010) 3803–3810. DOI:10.1016/j.ejmech.2010.05.030 |
| [9] | P. Barraja, P. Diana, A. Montalbano, et al., Pyrrolo[3, 4-h]quinolinones a new class of photochemothera peutic agents. Bioorg. Med. Chem. 19 (2011) 2326–2341. DOI:10.1016/j.bmc.2011.02.023 |
| [10] | K. Kaur, M. Jain, R.P. Reddy, R. Jain. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 45 (2010) 3245–3264. DOI:10.1016/j.ejmech.2010.04.011 |
| [11] | A. Marella, O.P. Tanwar, R. Saha, et al., Quinoline: a versatile heterocyclic. Saudi Pharm. J. 21 (2013) 1–12. DOI:10.1016/j.jsps.2012.03.002 |
| [12] | O. Afzal, S. Kumar, M.R. Haider, et al., A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 97 (2015) 871–910. DOI:10.1016/j.ejmech.2014.07.044 |
| [13] | R. Musiol, J. Jampilek, V. Buchta, et al., Antifungal properties of new series of quinoline derivatives. Bioorg. Med. Chem. 14 (2006) 3592–3598. DOI:10.1016/j.bmc.2006.01.016 |
| [14] | P. Palit, P. Paira, A. Hazra, et al., Phase transfer catalyzed synthesis of bisquinolines: antileishmanial activity in experimental visceral leishmaniasis and in vitro antibacterial evaluation. Eur. J. Med. Chem. 44 (2009) 845–853. DOI:10.1016/j.ejmech.2008.04.014 |
| [15] | R.S. Keri, S.A. Patil. Quinoline: a promising antitubercular target. Biomed. Pharmacother. 68 (2014) 1161–1175. DOI:10.1016/j.biopha.2014.10.007 |
| [16] | R.R. Soares, J.M.F.D. Silva, B.C. Carlos, et al., New quinoline derivatives demonstrate a promising antimalarial activity against Plasmodium falciparum in vitro and Plasmodium berghei in vivo. Bioorg. Med. Chem. Lett. 25 (2015) 2308–2313. DOI:10.1016/j.bmcl.2015.04.014 |
| [17] | S. Rasoul-Amini, A. Khalaj, A. Shafiee, et al., Anti-tumor activity of new quinoline derivatives in human breast cancer T47D cells. Int. J. Cancer Res. 2 (2006) 102–108. DOI:10.3923/ijcr.2006.102.108 |
| [18] | S.H. Chan, C.H. Chui, S.W. Chan, et al., Synthesis of 8-ydroxyquinoline derivatives as novel antitumor agents. ACS Med. Chem. Lett. 4 (2013) 170–174. DOI:10.1021/ml300238z |
| [19] | I.H.Y. Pun, D. Chan, S.H. Chan, et al., Anti-cancer effects of a novel quinoline derivative 83b1 on human esophageal squamous cell carcinoma (ESCC) through down-regulation of COX-2 mRNA and PGE2. Cancer Res. Treat. 18 (2016) . DOI:10.4143/crt.2016.190 |
| [20] | A. Mucha, P. Kafarski, Ł. Berlicki. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 54 (2011) 5955–5980. DOI:10.1021/jm200587f |
| [21] | W. Han, P. Mayer, A.R. Ofial. Iron-catalyzed oxidative mono-and bisphosphonation of NN-dialkylanilines. Adv. Synth. Catal. 352 (2010) 1667–1676. DOI:10.1002/adsc.v352:10 |
| [22] | P. Kafarski, B. Lejczak. Aminophosphonic acids of potential medical importance. Curr. Med. Chem. 1 (2001) 301–312. |
| [23] | S.M. Agawane, J.M. Nagarkar. Nano ceria catalyzed synthesis of α-aminophosphonates under ultrasonication. Tetrahedron Lett. 52 (2011) 3499–3504. DOI:10.1016/j.tetlet.2011.04.112 |
| [24] | L. Pan, X.H. Liu, Y.X. Shi, B.L. Wang, S.H. Wang. Solvent-and catalyst-free synthesis and antifungal activities of α-aminophosphonate containing cyclopropane moiety. Chem. Res. Chin. Univ. 26 (2010) 389–393. |
| [25] | S.A. Dake, D.S. Raut, K.R. Kharat, et al., Ionic liquid promoted synthesis, antibacterial and in vitro antiproliferative activity of novel α-aminophosphonate derivatives. Bioorg. Med. Chem. Lett. 21 (2011) 2527–2532. DOI:10.1016/j.bmcl.2011.02.039 |
| [26] | G.Y. Yao, M.Y. Ye, R.Z. Huang, et al., Synthesis and antitumor activities of novel rhein (-aminophosphonates conjugates. Bioorg. Med. Chem. Lett. 24 (2014) 501–507. DOI:10.1016/j.bmcl.2013.12.030 |
| [27] | G.S. Reddy, K.U.M. Rao, C.S. Sundar, et al., Neat synthesis and antioxidant activity of α-aminophosphonates. Arab. J. Chem. 7 (2014) 833–838. DOI:10.1016/j.arabjc.2013.01.004 |
| [28] | N. Gangwar, V.K. Kasana. Tartaric acid-catalyzed synthesis of α-aminophosphonates under solvent-free conditions. Synth. Commun. 41 (2011) 2800–2804. DOI:10.1080/00397911.2010.515358 |
| [29] | M.Y. Ye, G.Y. Yao, J.C. Wei, et al., Synthesis, cytotoxicity DNA binding and apoptosis of rhein-phosphonate derivatives as antitumor agents. Int. J. Mol. Sci. 14 (2013) 9424–9439. DOI:10.3390/ijms14059424 |
| [30] | X.C. Huang, M. Wang, Y.M. Pan, et al., Synthesis and antitumor activities of novel α-aminophosphonates dehydroabietic acid derivatives. Bioorg. Med. Chem. Lett. 23 (2013) 5283–5289. DOI:10.1016/j.bmcl.2013.08.005 |
| [31] | Y.C. Guo, J. Li, J.L. Ma, et al., Synthesis and antitumor activity of α-aminophosphonate derivatives containing thieno[2, 3-d]pyrimidines. Chin. Chem. Lett. 26 (2015) 755–758. DOI:10.1016/j.cclet.2015.03.026 |
| [32] | S.K. Kim, W.P. Ding, L. Zhang, W. Tian, S.Y. Chen. Clinical response to sunitinib as a multitargeted tyrosine-kinase inhibitor (TKI) in solid cancers: a review of clinical trials. Onco Targets Ther. 7 (2014) 719–728. |
| [33] | D. Koeberle, M. Montemurro, P. Samaras, et al., Continuous Sunitinib treatment in patients with advanced hepatocellular carcinoma: a Swiss group for clinical cancer research (SAKK) and Swiss association for the study of the liver (SASL) multicenter phase Ⅱ trial (SAKK 77/06). Oncologist 15 (2010) 285–292. DOI:10.1634/theoncologist.2009-0316 |
| [34] | L.L. Zhang, E. Meggers. An extremely stable and orthogonal DNA base pair with a simplified three-carbon backbone. J. Am. Chem. Soc. 127 (2005) 74–75. DOI:10.1021/ja043904j |