 2017, Vol. 28
2017, Vol. 28 



Decarboxylative bromination of the carboxylic acids is useful in organic chemistry to obtain bromo-compounds [1]. A number of metal catalysts including mercury, lithium, lead, manganese, silver [2-6] and non-metallic hypervant iodine [7, 8] were employed to access these bromo-compounds. Besides, decarboxylative bromination catalyzed by NaNO2 in combination with HBr in the presence of oxygen was reported previously [9]. However, most of these reactions usually involved heavy metal salts with a little slow process although these methods are highly efficient. As a result, a green alternative methodology for decarboxylative bromination is still desirable.
As far as we know, there has been no report on decarboxylative bromination via an electrochemical oxidation yet. Our group have developed a series of electrochemical routes toward various organic transformations [10-16]. This encourages us to make use of electrochemistry to accomplish a decarboxylative bromination directly. Herein we report an electrochemical decarboxylative bromination of cinnamic acids in aqueous media. This method features transition-metal-free, shorter time and no additional supporting electrolyte.
2. Results and discussionInitially, the reaction of cinnamic acid 1a with sodium-bromide in an undivided cell equipped with a pair of C—C electrodes. The mixture was electrolyzed under a constant current of 40 mA with continuous stirring at room temperature for 1 h. Among various solvents examined (Table 1, entries 1-6), MeCN/H2O (7 mL/3 mL) gave the best yield to 37% (Table 1, entry 1). Testing of the ratio of solvent and H2O showed the best ratio is 7/3 (entries 1, 7, 8). Next, we optimized the reaction temperature from 0 ℃ to 90 ℃ (entries 9-12). Also, the electrode matches (Table 1, entries 13-15) and the current (entries 16-17) were optimized, and the result showed that better yields could be obtained when the reaction was set up by using a platinum plate anode and a platinum plate cathode under a constant current of 40 mA. The optimization of the bromine source indicated that ammonium-bromide was the best choice (entries 18-20). In addition, we increased the loading of NH4Br to 6 equiv., giving 3a in 67% yield (Table 1, entry 21). However, yield of the reaction decreased when Et3N or AcOH was added to the reaction mixture (Table 1, entries 22-23). Finally, the reaction was set up as follows: cinnamic acid 1a (0.5 mmol), ammonium-bromine 2 (3 mmol) and MeCN/H2O (7 mL/3 mL) as the solvent in an undivided cell, which equipped with platinum anode and cathode, the reaction being carried out at 75 ℃ under a constant current of 40 mA for 1 h.
|
|
Table 1 Optimization of the reaction conditions.a |
Under the optimized conditions, different substituents at the phenyl ring of cinnamic acids were examined in the reaction (Table 2). The cinnamic acid bearing electron-donating groups (4-CH3, 4-OCH3) on the phenyl ring gave the desired products (3b, 3c) in higher yields than those bearing electron-withdrawing groups (4-Cl, 4-Br), as shown in 3d, 3e. And, 4-(trifluoromethyl) cinnamic acid, a strong electron-deficient cinnamic acid, gave a trace amount of the desired product 3f. In contrast, the strong electron-donation group favored the reaction. For instance, (E)-5-(2-bromovinyl)-1, 2, 3-trimethoxybenzene (3n) was obtained with a high yield of 83%. The investigation indicated that the effect of steric hindrance had a negative influence on the reaction. For instance, the o-substituted products could be obtained with low yields of 42%-63% (3j, 3k, 3l, 3m). Similarly, the m-substituted substrates gave the corresponding products (3g, 3h, 3i) in a little lower yields than the p-substituted cinnamic acids (3b, 3c, 3d). Notably, When R2 was phenyl, the desired product can be obtained with a high yield of 86% (3o). The phenyl ring can be replaced with hetero-aromatic or naphthalic ring. For example, 3-(2-thienyl) acrylic acid can be employed as the substrate, affording the desired bromo-product in spite of a low yield of 32% (3p). Similarly, only 28% yield of (E)-1-(2-bromovinyl)naphthalene (3q) can be obtained in the reaction. These lower yields perhaps can be ascribed to the electrochemical instability of thiophene ring and the strong steric effect of the naphthalic ring. On the other hand, the substrates of crotonic acid or phenylpropiolic acid hardly gave the desired product in the reaction, perhaps due to the instability of the corresponding intermediate F in mechanism.
|
|
Table 2 The scope for the decarboxylative bromination.a |
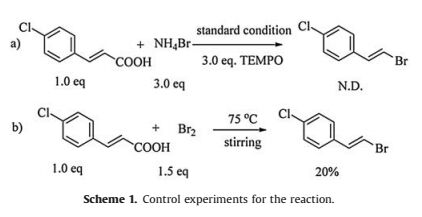
To gain an insight into the reaction mechanism, the process of the reaction were studied (Scheme 1). First, it was observed that the reaction was inhibited in the presence of 3.0 equiv. of the 2, 2, 6, 6-tetramethyl-1-piperidiny-loxy (TEMPO). Nevertheless, no radical was trapped when 5, 5-dimethyl-1-proline-N-oxide (DMPO) was added to the reaction in the electron paramagnetic resonance (EPR) experiments. Therefore, a molecular bromine process was proposed. As expected, the corresponding product could be obtained when NH4Br was replaced by Br2 without electrolysis.

|
Download:
|
| Scheme1. Control experiments for the reaction. | |
{kind=link}
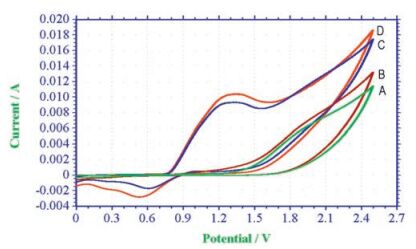
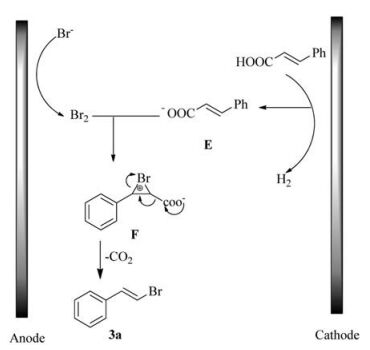
At the same time, the cyclic voltammetry (CV) experiments were performed to investigate the reaction process (showed in Fig. 1). Curve A and B showed that cinnamic acid was not oxidized in the potential range 0.0-2.5 V vs. Ag/AgCl. Curve C exhibited one oxidation wave at 1.32 V. We assigned the peak to the oxidation from Br- to produce molecular bromine [17, 18]. Actually, the color of the cell solution turned to be yellow when this oxidation peak appeared. Curve D showed that a slightly increase in the catalytic current was observed when 1a was added to curve C. (Note the conversion of E to F in Scheme 2.)

|
Download:
|
| Fig. 1. Cyclic voltammogram curves of cinnamic acid and NH4Br in 0.1 mol/L LiClO4/ CH3CN/H2O = 7:3 (10 mL), using platinum plate as working electrode, Pt wire and Ag/AgCl as counter and reference electrode at 50 mV/s scan rate. (A, 0.1 mol/L LiClO4. B, 2 mmol/L cinnamic acid and 0.1 mol/L LiClO4. C, 12 mmol/L NH4Br and 0.1 mol/L LiClO4. D, 2 mmol/L cinnamic acid, 12 mmol/L NH4Br and 0.1 mol/L LiClO4.). | |
{kind=link}

|
Download:
|
| Scheme2. Proposed mechanism for the decarboxylative bromination. | |
{kind=link}
Based on above experiments and literature reports [19], a plausible mechanism is proposed and is shown in Scheme 2. The anodic oxidation of bromide ion generates molecular bromine, which attack anion E to obtain the intermediate F. Then intermediate F is decarboxylated to afford the bromide 3a.
3. ConclusionIn conclusion, a new decarboxylative bromination of α, β-unsaturated carboxylic acids was developed by a direct anodic oxidation in the presence of ammonium-bromine. And a tentative mechanism was proposed on the base of the control experiments and the cyclic voltammetry (CV) experiments. Further investigations to develop decarboxylative chlorination of α, β-unsaturated carboxylic acids by using modified electrode [20, 21] are underway in our laboratory.
4. Experimental sectionThe instrument for electrolysis is dual display potentiostat (DJS-292) (made in China). 1H NMR and 13C NMR were recorded on a Bruker AC-300 FT (1H: 400 MHz, 13C: 100 MHz) using TMS as internal reference. Commercial reagent and compound were used without purification unless otherwise indicated. β-Phenyl substituted cinnamic acid was prepared according to literature procedures [22].
Substrate 1 (0.5 mmol), NH4Br (3 mmol) were added to an undivided cell which equipped with platinum anode and cathode. Followed by addition of MeCN (7 mL) and H2O (3 mL). The mixture was electrolyzed under continuous stirring at 75 ℃ under a constant current of 40 mA for 1 h. The solution was cooled to r.t., condensed under vacuum, extracted with DCM (3 × 10 mL), washed with saturated sodium chloride solution. And the organic layers were dried over Na2SO4, filtered and evaporated under vacuum. The residue was purified by column chromatography on silica gel (petroleum ether:ethyl acetate = 50:1-30:1) to afford the desired product 3.
Characterization data of compounds 3 were given in Supporting information.
AcknowledgmentsThe authors are grateful to the National Nature Science Foundation of China (Nos. 21472177, 21672200, 21432009, J1310010) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB20000000).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2017.04.030.
| [1] | H. Hunsdiecker, C. Hunsdiecker. Über den Abbau der Salzealiphatischer Säurendurch Brom. Ber 75(1942)291–297. |
| [2] | S. Cristol, Jr W. Firth. A convenient synthesis of alkyl halides from carboxylic acids. J. Org. Chem. 26(1961)280. DOI:10.1021/jo01060a628 |
| [3] | D. Naskar, S. Roy. Synthesis of α-bromo-β-lactam via a novel catalytic Hunsdiecker like protocol. J. Chem. Soc. Perkin Trans. 1(1999)2435–2436. |
| [4] | S. Khamarui, D. Sarkar, P. Pandit, D.K. Maiti. A fast and selective decarboxylativ edifunctionalization and cyclization for easy access to gem-dihalo alcohol ether. ester and bromo-1, 4-dioxane, Chem. Commun. 47(2011)12667–12669. |
| [5] | C.X. Kuang, H. Senboku, M. Tokuda. Stereoselective synthesis of (E)-b-arylvinyl bromides by microwave-induced reaction of anti-3-aryl-2.3-dibromopropanoic acids using an AgOAc-AcOH system. Tetrahedron 61(2005)637–642. DOI:10.1016/j.tet.2004.10.102 |
| [6] | R.A. Sheldon, J.K. Kochi. Oxidative decarboxylation of acids by lead tetraacetate. J. Org. React. 19(1972)275–278. |
| [7] | J.P. Das, S. Roy. Catalytic Hunsdiecker reaction of α.β-unsaturated carboxylic acids:how efficient is the catalyst?,. J. Org. Chem. 67(2002)7861–7864. DOI:10.1021/jo025868h |
| [8] | A. Graven, K.A. Joergensen, S. Dahl, A. Stanczak. Oxidative halodecarboxylation of α.β-unsaturated carboxylic acids. J. Org. Chem. 59(1994)3543–3546. DOI:10.1021/jo00092a009 |
| [9] | V.N. Telvekar, B.S. Takale. A novel method for bromo-decarboxylation of α.β-unsaturated carboxylic acids using catalytic sodium nitrite. Tetrahedron Lett. 52(2011)2394–2396. DOI:10.1016/j.tetlet.2011.02.101 |
| [10] | L. Zhang, H. Chen, Z.G. Zha, Z.Y. Wang. Electrochemical tandem synthesis of oximes from alcohols using KNO3 as the nitrogen source. mediated by tin microspheres in aqueous medium. Chem. Commun. 48(2012)6574–6576. DOI:10.1039/c2cc32800c |
| [11] | L. Meng, J.H. Su, Z.G. Zha, L. Zhang, Z.Y. Wang. Direct electrosynthesis of ketones from benzylicmethylenes by electrooxidative C-H activation. Chem. Eur. J. 19(2013)5542–5545. DOI:10.1002/chem.v19.18 |
| [12] | Z.L. Zhang, J.H. Su, Z.G. Zha, Z.Y. Wang. Electrochemical synthesis of the aryl α-ketoesters from acetophenones mediated by KI. Chem. Eur. J. 19(2013)17711–17714. DOI:10.1002/chem.201302307 |
| [13] | J.Q. Ye, Z.L. Zhang, Z.G. Zha, Z.Y. Wang. A green and efficient access to aryl nitriles via an electrochemical anodic oxidation. Chin.Chem. Lett. 25(2014)1112–1114. DOI:10.1016/j.cclet.2014.04.024 |
| [14] | H.H. Gao, Z.G. Zha, Z.L. Zhang, H.Y. Ma, Z.Y. Wang. A simple and efficient approach to realize difunctionalization of arylketones withmalonate estersvia an electrochemical oxidation. Chem. Commun. 50(2014)5034–5036. DOI:10.1039/C4CC01277A |
| [15] | K. Xu, Z.L. Zhang, P. Qian, Z.G. Zha, Z.Y. Wang. Electrosynthesis of enaminones directly from methyl ketones and amines with nitromethane as a carbon source. Chem. Commun. 51(2015)11108–11111. DOI:10.1039/C5CC02730F |
| [16] | P. Qian, M.X. Bi, J.H. Su, Z.G. Zha, Z.Y. Wang. Electrosynthesis of (E)-vinyl sulfonesdirectly from cinnamicacids and sodium sulfinates via decarboxylative sulfonofunctionalization. J. Org. Chem. 81(2016)4876–4882. DOI:10.1021/acs.joc.6b00661 |
| [17] | G.D. Allen, M.C. Buzzeo, C. Villagra'n, C. Hardacre, R.G. Compton, A mechanistic studyofthe electro-oxidation of bromide in acetonitrile and the room temperature ionic liquid, 1-butyl-3-methylimidazolium bis (trifluoromethylsulfonyl)imide at platinum electrodes, J. Electroanal. Chem. 575(2005) 311-320. |
| [18] | I. Damljanovic', D. Stevanovic', M. Vukic'evic', R.D. Vukic'evic, Electrochemical bromochlorination of peracetylated glycols, Carbohydr. Res. 346(2011) 2683-2687. |
| [19] | L.S. Kang, M.H. Luo, C.M. Lam, et al., Electrochemical C-H functionalization and subsequent C-S and C-N bond formation:Paired electrosynthesis of 3-amino-2-thiocyanatoa, α.β-unsaturated carbonylderivatives mediated by bromide ion, Green Chem. 18(2016) 3767-3774. |
| [20] | C.W. Zhang, L.B. Xu, J.F. Chen. High loading Pt nanoparticles on ordered mesoporous carbon spherearrays for high lyactive methanol electrooxidation. Chin. Chem. Lett. 27(2016)832–836. DOI:10.1016/j.cclet.2016.02.025 |
| [21] | N. Rahbar, Z. Ramezani, J. Ghanavati. CuO-nanoparticles modified carbon paste electrode or square wave voltammetric determination of lidocaine:Comparing classical and Box-Behnken optimization methodologies. Chin. Chem. Lett. 27(2016)837–842. DOI:10.1016/j.cclet.2016.04.017 |
| [22] | R.Q. Wu, M.G. Beauchamps, J.M. Laquidara, J.R. Sowa Jr. Ruthenium-catalyzed asymmetric transfer hydrogenation of allylic alcohols by an enantioselective isomerization/transfer hydrogenation mechanism. Angew. Chem. Int. Ed. 51(2012)2106–2110. DOI:10.1002/anie.201107910 |