 2017, Vol. 28
2017, Vol. 28 



b College of Chemistry and Materials Engineering, Quzhou University, Quzhou 324000, China
Optically pure β-arylmethyl-β-amino acids show very extensive application in life science as fundamental moieties of biologically active peptides and small-molecular drugs [1]. For example, a matrix metalloproteinase-2 inhibitor β-tetrapeptide has (S) -β-benzyl-β-amino acid units [2]. As first dipetidyl peptidase IV inhibitor approved by USFDA in 2006, sitagliptin phosphate (Januvia) contains a (R) -3-amino-4- (2, 4, 5-trifluorophenyl) butanoic acid subunit [3]. Moreover, enantiopure β-arylmethyl-β-amino acids are common chiral building blocks for organic synthesis [4]. Generally, the formation of chiral center of enantiopure β-arylmethyl-β-amino acids is based on as follows: (1) asymmetric hydrogenation of an enamine via expensive ligands and toxic Pt, Rh or Ru metal [5]; (2) homologation of an enantiopure α-amnio acid by the Arndt-Eistert method [6] or by reduction to α-amnio alcohol, which is converted to the corresponding cyanide, followed by hydrolysis [7]; (3) enzymatic catalyzed hydrolysis and alcoholysis of racemic β-lactam rings or β-amino esters [8]; (4) the reduction and substitution of enantiomeric lactone prepared from optically pure aspartic acid [9]; (5) conjugate addition of chiral amine to α, β-unsaturated ester [10] or conjugate addition of amine to α, β-unsaturated ketone catalyzed with chiral Lewis acids [11]; (6) palladium-catalyzed cross-coupling reaction between enantiopure organozinic reagent derivated from α-amnio acids and aryl iodides [12]; (7) ring opening of enantiomeric β-lactone or β-lactam with various nucleophilic reagent [13]; Recently Zhou et al. developed chemical kinetic resolution of unprotected β-substituted β-amino acids using recyclable chiral ligands to obtain optically pure β-benzylb-amino acid and 3-amino-4- (2, 4, 5-trifluorophenyl) butanoic acid [14].
With the rapid development of asymmetric organocatalysis, a variety of hemiacetals can easily be prepared with high enantioselectivity via the secondary amine-catalyzed tandem conjugate addition between N-protected hydroxylamines and α, β-enals as key step [15]. During our preparation of manuscript, Dou et al. reported a spiro-pyrrolidine-catalyzed asymmetric conjugate addition of hydroxylamine to enals and 2, 4-dienals to form hemiacetals [16].
Sequential one-pot or cascade reaction can improve reaction efficiency and avoid the time-consuming purification of intermediates [17]. Catalyzed by commercially available Grubbs second-generation catalyst (Grubbs-II catalyst), the cross-metathesis (CM) of terminal olefins with crotonaldehyde can afford α, β-enals in high yield and high E/Z selectivity, which can subsequently react with a variety of nucleophilic reagent in a onepot fashion [18].
Herein we investigated asymmetric conjugate addition between N-protected hydroxylamines and (E) -4-phenylbut-2-enals catalyzed by both chiral secondary amine and Brønsted acid. Furthermore, various (3R, 5S) -N-Boc-3-arylmethyl-5-hydroxyisoxazolidines were synthesized via one-pot olefin cross-metathesis and conjugate addition. In the end, valuable chiral N-Boc-β-benzyl-β-amino acid was concisely synthesized in two-step short procedure via construction of chiral N-Boc-3-benzyl-5- oxoisoxazolidine through cross-Metathesis/conjugate addition/ oxidation starting from allybenzene and crotonaldehyde.
2. Results and discussionInitially, we tried the conjugate addition of (E) -4-phenylbut-2- enal 1 and N-Boc-protected hydroxylamine 2 or N-Cbz-protected hydroxylamine catalyzed by secondary amine catalyst (Fig. 1, I, II or III) without Brønsted acid additive followed by the reported literatures [15b]. However, we failed to get any products and the starting materials were recovered. We envisioned that hemiacetal formation via subsequent tandem intramolecular cyclization may drive the conjugate addition. On the other hand Brønsted acid can activate formyl group in the conjugate addition adduct to facilitate intramolecular hemiacetal formation.

|
Download:
|
| Figure 1. The structures of catalysts. | |
Thus, the influence of Brønsted acid additives was evaluated, and the results were shown in Table 1. In the presence of 20 mol% piperidine 3a and 20 mol% benzoic acid (pKa = 4.19) [19] in chloroform, the conjugate addition of (E) -4-phenylbut-2-enal 1 and N-Cbz-protected hydroxylamine could not happen at room temperature and the starting materials were recovered. Further, we examined the reaction of (E) -4-phenylbut-2-enal 1 and N-Bocprotected hydroxylamine catalyzed by 20 mol% piperidine I and 20 mol% benzoic acid in chloroform at 0 ℃ (Table 1, entry 1). The intermolecular hemiacetal 5 was isolated in 91% yield. It seems that acidity of the reaction mixture in the presence of piperidine I and benzoic acid is not strong enough to activate the formyl group in the conjugate addition to in-situ form intramolecular hemiacetal product 3a. With the presence of 20 mol% (S) -diphenylprolinolTMS II together with 20 mol% benzoic acid as catalyst (entry 2), compound 3a and compound 5 were isolated in 71% yield with 89% ee and 11% yield, respectively. Fortunately, using 20 mol% pnitrobenzoic acid (pKa = 3.47) as additive, compound 4a was afforded in 84% with 92% ee (entry 3) at 0 ℃. Stronger carboxylic acids such as phosphoric acid (pKa = 2.12) and TFA (pKa = 0.23), TsOH (pKa = -2.8) promoted the in-situ formation of intramolecular hemiacetal in conjugate addition reaction (entries 4-6), however the enantioselectivities are low and double-Boc derivative 4 was isolated. Though stronger Brønsted acid (phosphoric acid and TFA, TsOH) can activate the formyl group in the conjugate addition adduct to in situ form intramolecular hemiacetal product, the migration of Boc group from N-Boc-protected hydroxylamine happened in this case.
|
|
Table 1 Conjugate addition between (E)-4-phenylbut-2-enal and N-protected hydroxylamines in the presence of secondary amine and Brønsted acid additives.a |

A solvent screening showed that the reactions catalyzed by (S) - diphenylprolinol-TMS II and p-nitrobenzoic acid generally proceeded smoothly in other solvents, such as Et2O, methanol, toluene and dichloromethane (DCM) (entries 7-10). Dichloromethane (DCM) afforded slightly higher yield. While the loading amount of (S) -diphenylprolinol-TMS II and p-nitrobenzoic acid were reduced to 10 mol%, no loss of yield and enenatioselectivity can be observed (entry 11). Next, we explored the effect of catalysts on the reaction in dichloromethane solvent. Calalyst III containing 3, 5-difluorophenyl groups instead of phenyl groups resulted in poor enantioselectivity (45% ee) (entry 12). Catalyst IV gave intramolecular hemiacetal 3a in low yield with poor enantioselectivity (entry 13).
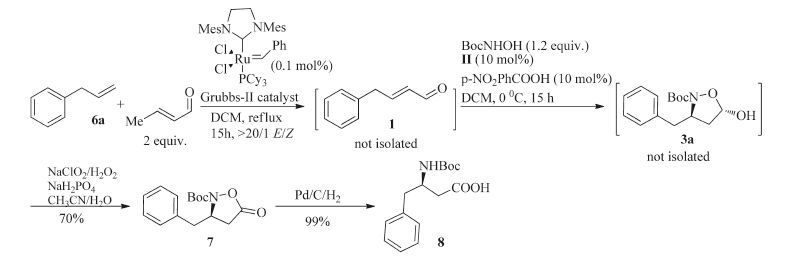
Then, the one-pot CM/conjugate addition was performed (Scheme 1). The reaction mixture of terminal olefin 6a and excess (2.0 equiv.) of crotonaldehyde together with 0.1 mol% Grubbs-II catalyst in dichloromethane was refluxed for 12 h. Checked by 1H NMR, the terminal olefin 6a was quantitatively converted to (E) -4- phenylbut-2-enal 1 (>20/1 E/Z). The loading amount of Grubbs-II catalyst can be reduced to 0.05 mol%, however the reaction was prolonged to 48 h. The above reaction mixture was cooled to 0 ℃, and then N-Boc-protected hydroxylamine 2 (1.2 equiv.), (S) - diphenylprolinol-TMS II (0.1 equiv.) and p-nitrobenzoic acid (0.1 equiv.) were subsequently added to it. The reaction mixture was stirred for 15 h and compound 3a was isolated in 86% yield with 92% ee.

|
Download:
|
| Scheme 1. One-pot CM/conjugate addition for the enantioselective synthesis of (3R,5S)-N-Boc-3-benzyl-5-hydroxyisoxazolidine. | |
With the optimized condition in hand, we further explored the substrate scope of the one-pot CM/conjugate addition (Table 2). In the presence of 10 mol% of (S) -diphenylprolinolTMS II and 10 mol% of p-nitrobenzoic acid in dichloromethane, the one-pot CM/conjugate addition gave the corresponding (3R) - tert-butyl 3-arylmethyl-5-hydroxyisoxazolidine-2-carboxylate 3 in moderate to good yields with good enantioselectivities (entries 1-10). The use of 2-allylnaphthalene 6g resulted in higher enantioselectivity (95% ee), which can be attributed from more bulky group adjacent to reacting center (entry 7). It was necessary to mention that 1-allyl-2, 4, 5-trifluorobenzene 6j gave corresponding 5-hydroxyisooxazolidines 3j, a potential intermediate for the synthesis of sitagliptin phosphate in 79% yield with 93% ee. It may be attributed to the high reactivity of multiple-fluoro phenyl group.
|
|
Table 2 Scope of one-pot CM/conjugate addition.a |

{kind=link}
{kind=link}
We then turned our attention to convert (3R, 5S) -N-Boc-3- benzyl-5-hydroxyisoxazolidine 3a to chiral β-benzyl-β-amino acid. Oxidation/NO bond cleavage sequence reaction is a common methodology to furnish this transformation. However, the conversion of compound 3a to isooxazolidin-5-one 7 did not take place under the condition of NaClO2/NaH2PO4/2-methyl-2-butene or by PDC oxidation. NaClO2/H2O2/NaH2PO4 oxidation was normally used in conversion of aldehydes to carboxylic acids [20]. When we tried to convert (3R, 5S) -N-Boc-3-benzyl-5-hydroxyisoxazolidine 3a to (R) -N-Boc-3-benzyl-5-oxoisoxazolidine 7 with NaClO2/H2O2/NaH2PO4 oxidation, fortunately (R) -N-Boc-3- benzyl-5-oxoisoxazolidine 7 was obtained in 82% yield without the loss of enantioselectivity (Scheme 2). Finally, chiral (R) -N-Boc β-benzyl-β-amino acid 8 was obtained after hydrogenation in 99% yield. The absolute configuration of 8 was unambiguously revealed as R ([α]D25 + 18.1 (c 0.67 in MeOH); Ref. [21] [α]D25 + 19.7 (c 0.67 in MeOH) ). Furthermore, chiral (R) -N-Boc-β-benzyl-β-amino acid 8 was synthesized in 69% total yield in two-step procedure starting from allylbenzene 6a and crotonaldehyde (Scheme 3). With this methodology, multi-gram of chiral (R) -N-Boc-β-benzyl-β-amino acid 8 was obtained in 70% total yield.

|
Download:
|
| Scheme 2. Conversion of (3R,5S)-N-Boc-3-benzyl-5-hydroxyisoxazolidine 3a. | |
{kind=link}

|
Download:
|
| Scheme 3. Concise enantioselective synthesis of β-benzyl-β-amino acid in two-step procedure. | |
{kind=link}
3. Conclusion
In summary, we reported a concise enantioselective synthesis of valuable chiral N-Boc-β-benzyl-β-amino acid in two-step procedure. The synthetic strategy is based on the rapid construction of chiral N-Boc-3-benzyl-5-oxoisoxazolidine via cross-Metathesis/conjugate addition/oxidation. All of the starting materials for the synthesis of chiral N-Boc-β-benzyl-β-amino acid are cheap and procedure can be scaled up for the preparation of various chiral β-aryl-β-amino acids and important drugs, such as sitagliptin phosphate.
4. ExperimentalAll the reactions were monitored by thin-layer chromatography (TLC) that was performed on silica gel plates GF254. Visualization was achieved under a UV lamp (254 nm and 365 nm), and by developing the plates with potassium permanganate in water. Flash chromatography was performed using silica gel (200- 300 mesh) with solvents indicated in the text. NMR spectra were registered in a Bruker Advance 400 Ultrashield spectrometer in CDCl3 at room temperature, operating at 400 MHz (1H) and 100 MHz (13C). Chemical shifts are reported in ppm. High performance liquid chromatography (HPLC) was performed on a Shimadzu chromatograph (Essentia LC-16), using Chiralcel columns and guard columns. The spectral data of all the compounds are presented in the Supporting information.
Procedure for concise enantioselective synthesis of valuable N-Boc-β-benzyl-β-amino acid in two-step procedure: A solution of allylbenzene 6a (1.18 g, 10 mmol) and crotonaldehyde (1.4 g, 20 mmol) together with 0.1 mol% Grubbs-II catalyst (9 mg, 0.01 mmol) in dichloromethane (100 mL) was refluxed for 12 h, and then it was cooled to 0 ℃. To the reaction mixture was subsequently added N-Boc-hydroxylamine 2 (1.59 g, 12 mmol) and catalyst II (330 mg, 1 mmol, 10 mol%) and p-nitrobenzoic acid (160 mg, 1 mmol, 10 mol%), and then the resulting solution was stirred at 0 ℃ for 15 h. The resulting solution was evaporated to remove the dichloromethane, and then acetonitile (50 mL), NaH2PO4 (240 mg, 2 mmol) in water (10 mL) and H2O2 (35 wt% in water, 1.4 mL, 14 mmol) was added to the residue. A solution of NaClO2 (1.27 g, 14 mmol) in water (14 mL) was dropwise added to the above reaction mixture at 10 ℃. The mixture solution was diluted with dichloromethane (200 mL) and washed with saturated sodium bicarbonate aqueous solution (50 mL) and brine (50 mL). The resulting organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified over silica gel column chromatography (PE/ EtOAc, 5:1-3:1) to give (R) -N-Boc-3-benzyl-5-oxoisoxazolidine 7 (1.98 g, 71% yield) as a white solid. 92% ee; Chiral HPLC condition: SHIMADZU Essentia LC-16 HPLC, Chiralcel AD-H column (250 × 4.6 mm, i.d.) with a mixture of hexane and 2-propanol (95:5) at a flow rate of 1.2 mL/min as the mobile phase, oven temperature was 28 ℃, 210 nm, tminor = 14.91 min, tmajor = 11.82 min.
To a solution of (R) -N-Boc-3-benzyl-5-oxoisoxazolidine 7 (1.98 g, 7.1 mmol, 92% ee) in MeOH (50 mL) was added Pd/C (20% w/w, 550 mg). The reaction mixture was hydrogenated under 90 atm for 24 h. The reaction mixture was filtered through Celite with ethyl acetate and concentrated to give (R) -N-Boc-β-benzylb-amino acid 8 (1.96 g, 99% yield) as a white solid. [α]D22 + 18.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) : δ 7.33-7.24 (m, 5H), 5.04- 4.97 (m, 1H), 2.98 (dd, 1H, J = 8 Hz, 16 Hz), 2.87-2.84 (m, 2H), 2.62 (dd, 1H, J = 4 Hz, 16 Hz), 1.30 (s, 9H).
AcknowledgmentWe acknowledge the financial support for this study from the National Natural Science Foundation of China (No. 21472110).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.10.005.
| [1] | E. Juaristi, V.A. Soloshonok, Enantioselective Synthesis of β-Amino Acids, 2nd edn., Wiley-VCH, New York, 2005. |
| [2] | T. Mukai, N. Suganuma, K. Soejima, Synthesis of β-tetrapeptide analog as a mother compound for the development of matrix metalloproteinase-2-imaging agents. Chem. Pharm. Bull. 56 (2008) 260–265. DOI:10.1248/cpb.56.260 |
| [3] | (a) A.E. Weber, Dipeptidylpeptidase IV inhibitors for the treatmentof diabetes, J. Med. Chem. 47(2004) 4135-4141; (b) S.H. Havale, M. Pal, Medicinal chemistry approaches to the inhibition of dipeptidyl peptidase-4 for the treatment of type 2 diabetes, Bioorg. Med. Chem. 17(2009) 1783-1802; (c) N.A. Thornberry, A.E. Weber, Discovery of JANUVIATM (Sitagliptin), a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes, Curr. Top. Med. Chem. 7(2007) 557-568; (d) C.H.S. Mclntosh, Dipeptidyl peptidase IV inhibitors and diabetes therapy, Front. Biosci. 13(2008) 1753-1773; (e) A.A. Desai, Sitagliptin manufacture:a compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis, Angew. Chem. Int. Ed. 50(2011) 1974-1976. |
| [4] | (a) R.P.S. Cheng, H. Gellman, W.F. DeGrado, β-Peptides:from structure to function, Chem. Rev. 101(2001) 3219-3232; (b) D. Seebach, J. Gardiner, β-Peptidic peptidomimetics, Acc. Chem. Res. 41(2008) 1366-1375. |
| [5] | (a) K.B. Hansen, J. Balsells, S. Dreher, et al., First generation process for the preparation of the DPP-IV inhibitor sitagliptin, Org. Process Res. Dev. 9(2005) 634-639; (b) K.B. Hansen, Y. Hsiao, F. Xu, et al., Highly efficient asymmetric synthesis of sitagliptin, J. Am. Chem. Soc. 131(2009) 8798-8804; (c) D. Steinhuebel, Y. Sun, K. Matsumura, et al., Direct asymmetric reductive amination, J. Am. Chem. Soc. 131(2009) 11316-11317. |
| [6] | (a) J. Podlech, D. Seebach, The Arndt-Eistert reaction in peptide chemistry:a facile access to homopeptides, Angew. Chem. Int. Ed. 34(1995) 471-472; (b) G. Guichard, S. Abele, D. Seebach, Preparation of N-fmoc-protected β2-and β3-amino acids and their use as building blocks for the solid-phase synthesis of β-peptides, Helv. Chim. Acta 81(1998) 187-206. |
| [7] | (a) P.W. Sutton, A. Bradley, M.R. Elsegood, et al., Design and synthesis of a novel cyclo-β-tetrapeptide, Tetrahedron Lett. 40(1999) 2629-2632; (b) J. Farras,X. Ginesta, P.W. Sutton,et al., β3-Amino acids bynucleophilic ringopening of N-nosyl aziridines, Tetrahedron 57(2001) 7665-7674. |
| [8] | (a) X.G. Li, T.K. Kanerva, Lipase-involved strategy to the enantiomers of 4-benzyl-β-lactamas a key intermediate in the preparation of β-phenylalanine derivatives, Adv. Synth. Catal. 348(2006) 197-205; (b) J. Kaman, M. Forro, F. Fulop, Enzyme-catalysed kinetic resolution of N,Odiacetyl derivatives of cyclic 1,3-amino alcohols, Tetrahedron:Asymmetry 12(2001) 1881-1886; (c) C. Palomo, M. Oiarbide, S. Bindi, A concise β-lactam route to short peptide segments containing β,β-disubstituted β-amino acids, J. Org. Chem. 63(1998) 2469-2474; (d) G. Tasnadi, E. Forro, F. Fulop, Improved enzymatic syntheses of valuable β-arylalkyl-β-amino acid enantiomers, Org. Biomol. Chem. 8(2010) 793-799. |
| [9] | (a) C.W. Jefford, J.B. Wang, An enantiospecific synthesis of β-amino acids, Tetrahedron Lett. 34(1993) 1111-1114; (b) C.W. Jefford, J. Mcnulty, Z.H. Lu, et al., The enantioselective synthesis of β-amino acids, their α-hydroxy derivatives, and the N-terminal components of bestatin and microginin, Helv. Chim. Acta 79(1996) 1203-1216. |
| [10] | C. Miniejew, F. Outurquin, X. Pannecoucke, Diastereoselective synthesis of aziridine esters via amino selanyl esters. Tetrahedron 62 (2006) 2657–2670. DOI:10.1016/j.tet.2005.12.030 |
| [11] | C. Palomo, M. Oiarbide, R. Halder, Catalytic enantioselective conjugate addition of carbamates. J. Am. Chem. Soc. 126 (2004) 9188–9189. DOI:10.1021/ja047004e |
| [12] | R.F. Jackson, I. Rilatt, P.J. Murray, The rate of elimination of a β-amino zinc reagent is reduced by using a better leaving group. Chem. Commun. (2003) 1242–1243. |
| [13] | (a) S.G. Nelson, K.L. Spencer, W.S. Cheung, et al., Divergent reaction pathways in amine additions to β-lactone electrophiles. An application to β-peptide synthesis, Tetrahedron 58(2002) 7081-7091; (b) K.B. Hansen, J. Balsells, S. Dreher, et al., First generation process for the preparation of the DPP-IV inhibitor sitagliptin, Org. Process Res. Dev. 9(2005) 634-639. |
| [14] | S.B. Zhou, J. Wang, X. Chen, Chemical kinetic resolution of unprotected β-substituted β-amino acids using recyclable chiralligands. Angew. Chem. Int. Ed. 53 (2014) 7883–7886. DOI:10.1002/anie.201403556 |
| [15] | (a) Y.K. Chen, M. Yoshida, D.W.C. Macmillanm, Enantioselective organocatalytic amine conjugate addition, J. Am. Chem. Soc. 128(2006) 9328-9329; (b) I. Ibrahem, R. Rios, J. Vesely, et al., Organocatalytic asymmetric 5-hydroxyisoxazolidine synthesis:a highly enantioselective route to b-amino acids, Chem. Commun. (2007) 849-851; (c) O.V. Maltsev, A.S. Kucherenko, A.L. Chimishkyan, et al., α,α-Diarylprolinolderived chiral ionic liquids:recoverable organocatalysts for the domino reaction between α,β-enals and N-protected hydroxylamines, Tetrahedron:Asymmetry 21(2010) 2659-2670; (d) G.L. Zhao, S.Z. Lin, A. Korotvicka, et al., Asymmetric synthesis of Maraviroc (UK-427857), Adv. Synth. Catal. 352(2010) 2291-2298. |
| [16] | Q.Y. Dou, Y.Q. Tu, Y. Zhang, Spiro-pyrrolidine-catalyzed asymmetric conjugate addition of hydroxylamine to enals and 2,4-dienals. Adv. Synth. Catal. 358 (2016) 874–879. DOI:10.1002/adsc.v358.6 |
| [17] | (a) L.F. Tietze, U. Beifuss, Sequential transformations in organic chemistry:a synthetic strategy with a future, Angew. Chem. Int. Ed. Engl. 32(1993) 131-163; (b) L.F. Tietze, Domino reactions in organic synthesis, Chem. Rev. 96(1996) 115-136; (c) A. Padwa, S.K. Bur, The domino way to heterocycles, Tetrahedron 63(2007) 5341-5378. |
| [18] | G. Sirasani, T. Paul, R.B. Andrade, Sequencing cross-metathesis and nonmetathesis reactions to rapidly access building blocks for synthesis. Tetrahedron 67 (2011) 2191–2205. |
| [19] | pKa values measured in water, in:D.D. Perrin, E.P. Serjeant, B. Dempsey (Eds.), pKa Predictions for Organic Acids and Bases, Champman and Hall, London, 1981. |
| [20] | E. Dalcanale, F. Montanari, Selective oxidation of aldehydes to carboxylic acids with sodium chlorite-hydrogen peroxide. J. Org. Chem. 51 (1986) 567–569. DOI:10.1021/jo00354a037 |
| [21] | M. Seki, K. Matsumoto, A novel approach to homochiral b-amino acids. Tetrahedron Lett. 37 (1996) 3165–3168. DOI:10.1016/0040-4039(96)00518-7 |