 2017, Vol. 28
2017, Vol. 28 


 , Gui-Min Xiaa, Xiu-Jun Liua, Yun Chaia, Hong-Wei Hea
, Gui-Min Xiaa, Xiu-Jun Liua, Yun Chaia, Hong-Wei Hea
b The First Affi liated Hospital of Zhengzhou University, Zhengzhou 450052, China
Cancers still remain the first and second leading causes of death in economically developed and developing countries, respectively [1, 2]. It is expected that annual cancer cases will rise from 14 million in 2012 to 22 million within the next two decades [3]. Encouragingly, substantial progress has been made to develop more effective and safer strategies for treating cancers, and many classes of antitumor agents with diverse novel structures have been introduced in the market or under development in the past decades [4]. However, there continues to be a need for new antitumor drugs from either novel scaffolds or further structural modifications of existing drugs.
Quinolones represent an extremely successful family of antibiotics that have a broad-spectrum antibacterial activity and relatively few side effects [5]. Among of them, ciprofloxacin, ofloxacin and levofloxacin are also used as second-line drugs to treat tuberculosis [6]. Recently, quinolones as "privileged building blocks" have been reported to display many "nonclassical" biological profiles, such as antitumor, anti-HIV-1 integrase, anti-HCV-NS3 helicase and -NS5B-polymerase activities [7, 8]. It's exciting that Voreloxin (Fig. 1) with a broad-spectrum antitumor activity [9] the first quinolone antitumor drug, was approved as an orphan drug for the treatment of acute myeloid leukemia by the US FDA in 2009, and phase I-III clinical trials in patients with various tumors are currently ongoing [10].

|
Download:
|
| Figure 1. Chemical structures of Voreloxin and 1a. | |
In the course of our search for more potent antitumor agents, we focused our interest on structural modifications of Voreloxin. Given that 3-aminopyrrolidine, but not (3S, 4S)-3-methoxy-4-(methylamino) pyrrolidine, is commercially available, compound 1a (Fig. 1) possessing the in vitro antitumor activity comparable to Voreloxin [11] was chosen as the lead compound in our work. First, the thiazoly-2-yl group at the N-1 position of 1a was replaced with its isosteres (1H-imidazol-2-yl and oxazol-2-yl) to examine whether it is the optimal group of this position. On the other hand, our previous works have emphasized the importance of an oxime functional moiety of the C-7 side chain with respect to biological activities of quinolones [12-15]. Thus, a four, five-or sixmembered nitrogen heterocycle containing an alkoxyimino group was introduced at the C-7 position instead of the 3-aminopyrrolidin-1-yl one of 1a, to identify if a nitrogen heterocycle with an oxime moiety is permitted at this position. Our primary objective was to optimize the potency of these compounds against human cancer cell lines. A simple structure-activity relationship (SAR) study was also explored to facilitate the further development of the naphthyridinone derivatives.
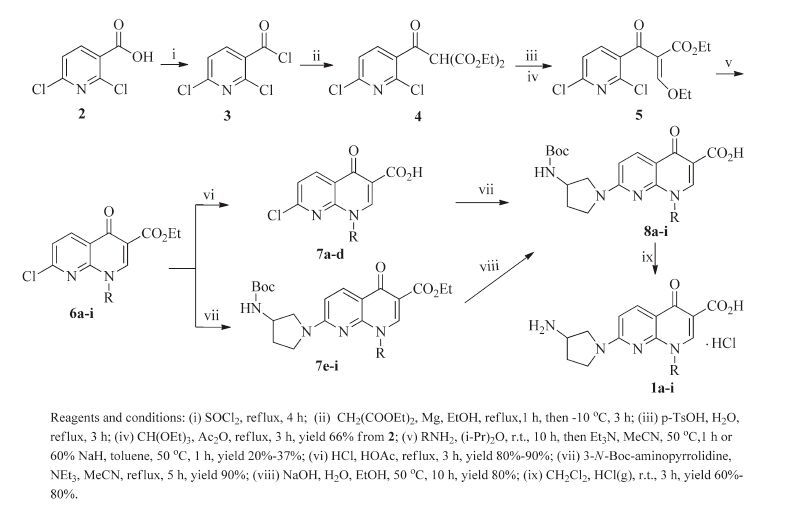
2. Experimental 2.1. Synthetic chemistryTo study the effect of various substituents at the N-1 position of the 7-(3-aminopyrrolidin-1-yl) naphthyridinone core, eight analogs of 1a were designed. A synthetic route to these compounds is illustrated in Scheme 1. The key intermediates 6a-i were readily prepared from 2, 6-dichloronicotinic acid (2) via a five-step procedure [14, 16]. Hydrolysis of the core esters 6a-d in HCl-HOAc followed by condensation with 3-N-Boc-aminopyrrolidine in the presence of triethylamine gave 8a-d. However, the esters 6e-i were not converted to the corresponding acids in a similar manner as for preparation of 8a-d. After various attempts, 8e-i were successfully obtained from 6e-i by condensation with the side chain compounds and then hydrolysis of the resulted esters 7e-i. The Boc-protecting group of 8a-i was removed by hydrogen chloride gas in dichloromethane to yield the desired naphthyridinone derivatives 1a-i as hydrochloric acid salts.

|
Download:
|
| Scheme 1. Synthesis of 7-(3-aminopyrrolidin-1-yl) naphthyridinone derivatives 1a-i. (See Table 1 for structures) | |
The synthetic route of novel 1-(thiazol-2-yl) naphthyridinones 10a-n containing nitrogen heterocycles with an alkoxyimino group was depicted in Scheme 2. Condensation of 6a with various side chain compounds [12, 17-19] and then hydrolysis of the resulted esters 9a-n yielded 10a-n.

|
Download:
|
| Scheme 2. Synthesis of 1-(thiazol-2-yl) naphthyridinone derivatives 10a-n. (See Table 1 for structures) | |
Since the oxime group can exist in the E or Z configuration, it was necessary to determine the geometries of all the oxime target compounds 10a-n. Preparing X-ray quality single crystals of any oxime intermediate or product met with no success in this study, but the oxime geometry would be expected to have the E-configuration according to the data in published papers [20, 21]. The concrete synthetic procedures, the physical characteristics, and 1H NMR for all the synthesized compounds are listed in Supporting information.
2.2. Anti-tumor activitiesAll the synthesized target compounds were preliminarily investigated for their in vitro activity against HL60 (leukemia) at the concentration of 30 μmol/L by SRB (Sulforhodamine B) assay [22]. And the compounds having >70% inhibition were subjected to IC50 (50% inhibition concentrations) determination against ten human cancer cell lines, including HL60, HepG2 (liver carcinoma), HCT-116 (colon cancer), A549 (lung adenocarcinoma), PANC-1 (pancreatic carcinoma), Hela (cervical cancer), DU145 (prostatic cancer), SKOV3 (ovarian carcinoma), MCF-7 (breast cancer) and MCF-7/DOX (Doxorubicin-resistant MCF-7) by SRB assay.
3. Results and discussionThe inhibitory effects of the 7-(3-aminopyrrolidin-1-yl) naphthyridinone derivatives 1a-i and 1-(thiazol-2-yl) naphthyridinone derivatives 10a-n were evaluated and listed in Tables 1 and 2. Unfortunately, contrary to our predictions, the results indicated that all the newly synthesized 7-(3-aminopyrrolidin-1-yl) naphthyridinone derivatives 1b-i (inhibition rates: 0-28%) were much less active than the reference 1a (75%, Table 1). Therefore, our modifications are not effective, and the thiazol-2-yl group is optimal for the N-1 position.
|
|
Table 1 Structures and in vitro activity of compounds 1a-i and 10a-n against HL60 at 30μmol/L. |

|
|
Table 2 in vitro activity of selected compounds against ten cell lines. |

{kind=link}
{kind=link}
{kind=link}
The IC50 values were compared with those of Etoposide and 1a (Table 2). The selected 1-(thiazol-2-yl) naphthyridinones have potent activity against these tested human cancer cell lines. All of them (IC50: < 0.5-49.23 μmol/L) are more active or comparable to Etoposide and 1a (IC50: 1.17 to >50 μmol/L) against HCT-116, A549, SKOV3 and MCF-7/DOX. Contrary to 1a, compounds 10h-j show significantly better activity against MCF-7/DOX (IC50: < 0.5 μmol/L) than MCF-7 (IC50: 5.57-31.04 μmol/L). Moreover, 10j was found to have a broad-spectrum activity (IC50: < 0.5-6.25 μmol/L) against all of the tested cell lines including Etoposide-and/or 1a -resistant ones, and is 1.3 to >100 fold more potent than those of the two references against these cell lines, except HL60 and HepG2.
According to the biological evaluation results shown in Tables 1 and 2, the antitumor activity of the naphthyridinone derivatives in this study depends on both of the groups at the N-1 and C-7 positions. The relative contribution of the heteroaromatic ring at the N-1 position to activity is as follows: thiazol-2-yl (1a) >> 1H-imidazol-2-yl (1f) >thiophen-3-yl (1d), and the others (1b, 1c, 1e, 1g-i) have no activity (Table 1).
On the other hand, 1-(thiazol-2-yl) naphthyridinones 10a-n generally exhibit in vitro activity (Table 1), which indicates that a nitrogen heterocycle with an oxime group is permitted at the C-7 position. Although a simple SAR is difficult to explore, the sizes of the heterocycle and the oxime group are important for the activity. For example, the activity imparted to the 7-(3-aminomethyl-3-methylpyrrolidin-1-yl) naphthyridinone ring by the alkyl group of the oxime moiety is as follows: benzyl > ethyl > methyl (10j vs.10i vs.10h), which suggests that simply increasing the lipophilicity could improve the activity (Table 2). Above all, thiazol-2-yl and 3-aminomethyl-4-benzyl oxyimino-3-methylpyrrolidin-1-yl groups are the most active at the N-1 and C-7 positions of naphthyridinone core, respectively.
4. ConclusionIn summary, structural modifications of compound 1a (a precursor of Voreloxin) at the N-1 position (various heteroaromatic rings instead of the thiazol-2-yl) and C-7 position (four-/ five-/six-membered nitrogen heterocyclic amine moieties with an oxime group instead of 3-aminopyrrolidin-1-yl one), respectively, were made in this study. Our results reveal that thiazol-2-yl and 3-aminomethyl-4-benzyloxyimino-3-methylpyrrolidin-1-yl groups are optimal at the N-1 and C-7 positions of naphthyridinone core, respectively. The most active compound 10j shows considerable broad-spectrum antitumor activity (IC50: < 0.5-6.25 μmol/L) against all of the tested cell lines including Etoposide-and/or 1a-resistant ones and is 1.3 to >100 fold more potent than those of the two references against these cell lines, except HL60 and HepG2.
AcknowledgmentThis work was supported by National Key Research and Development Program (No. 2016YFA0201500), the National S & T Major Special Project on Major New Drug Innovations (Nos. 2014ZX09507009-003, 2015ZX09102007) and NSFC (Nos. 81373267, 21502237).
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2016.07.024.
| [1] | K. Lv, L.L. Wang, X.B. Zhou, Synthesis and in vitro antitumor activity of 1-(3-dimethylamino) propyl indolin-2-one derivatives. Med. Chem. Res. 22 (2013) 1723–1729. DOI:10.1007/s00044-012-0170-3 |
| [2] | A. Jemal, F. Bray, M.M. Center, Global cancer statistics. Cancer J. Clin. 61 (2011) 69–90. DOI:10.3322/caac.v61:2 |
| [3] | B.W. Stewart, C.P. Wild, World Cancer Report 2014, World Health Organization. International Agency for Research on Cancer (2015) . |
| [4] | M.S. Kinch, An analysis of FDA-approved drugs for oncology. Drug Discov. Today 19 (2014) 1831–1835. DOI:10.1016/j.drudis.2014.08.007 |
| [5] | T.T. Zhang, W.Y. Shen, M.L. Liu, Synthesis, antimycobacterial and antibacterial activity of fluoroquinolone derivatives containing an 3-alkoxyimino-4-(cyclopropylanimo) methylpyrrolidine moiety. Eur. J. Med. Chem. 104 (2015) 73–85. DOI:10.1016/j.ejmech.2015.09.030 |
| [6] | A. Zumla, P. Nahid, S.T. Cole, Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 12 (2013) 388–404. DOI:10.1038/nrd4001 |
| [7] | A. Ahmed, M. Daneshtalab, Nonclassical biological activities of quinolone derivatives. J. Pharm. Pharm. Sci. 15 (2012) 52–72. |
| [8] | L.S. Feng, M.L. Liu, Advances of structural modifications and nonclassical biological activities of quinolones derivatives. J. Int. Pharm. Res. 37 (2010) 139–143. |
| [9] | U. Hoch, J. Lynch, Y. Sato, Voreloxin, formerly SNS-595, has potent activity against a broad panel of cancer cell lines and in vivo tumor models. Cancer Chemother. Pharmacol. 64 (2009) 53–65. DOI:10.1007/s00280-008-0850-3 |
| [10] | J.A. Abbas, R.K. Stuart, Vosaroxin:a novel antineoplastic quinolone. Expert Opin. Investig. Drugs 21 (2012) 1223–1233. DOI:10.1517/13543784.2012.699038 |
| [11] | Y. Tsuzuki, K. Tomita, K.I. Shibamori, Synthesis and structure-activity relationships of novel 7-substituted 1, 4-dihydro-4-oxo-1-(2-thiazolyl)-1, 8-naphthyridine-3-carboxylic acids as antitumor agents. Part 2. J. Med. Chem. 47 (2004) 2097–2109. DOI:10.1021/jm0304966 |
| [12] | K. Lv, Y.X. Sun, L.Y. Sun, Design, synthesis, and in vitro antibacterial activity of fluoroquinolone derivatives containing a chiral 3-(alkoxyimino)-2-(aminomethyl) azetidine moiety. ChemMedChem 7 (2012) 1230–1236. DOI:10.1002/cmdc.v7.7 |
| [13] | K. Lv, J.W. Wang, M.L. Liu, Synthesis and in vitro antibacterial activity of quinolone/naphthyridone derivatives containing 3-alkoxyimino-4-(methyl) aminopiperidine scaffolds. Bioorg. Med. Chem. Lett. 23 (2013) 1754–1759. DOI:10.1016/j.bmcl.2013.01.048 |
| [14] | H.M. Liu, J. Huang, J.Y. Wang, Synthesis, antimycobacterial and antibacterial evaluation of l-[(1R, 2S)-2-fluorocyclopropyl]fluoroquinolone derivatives containing an oxime functional moiety. Eur. J. Med. Chem. 86 (2014) 628–638. DOI:10.1016/j.ejmech.2014.09.029 |
| [15] | J. Huang, H.T. Liu, M.L. Liu, Synthesis, antimycobacterial and antibacterial activity of l-[(1R, 2S)-2-fluorocyclopropyl]naphthyridone derivatives containing an oxime-functionalized pyrrolidine moiety. Bioorg. Med. Chem. Lett. 25 (2015) 5058–5063. DOI:10.1016/j.bmcl.2015.10.027 |
| [16] | K. Tomita, Y. Tsuzuki, K.I. Shibamori, Synthesis and structure-activity relationships of novel 7-substituted 1, 4-dihydro-4-oxo-1-(2-thiazolyl))-1, 8-naphthyridine-3-carboxylic acids as antitumor agents. Part 1. J. Med. Chem. 45 (2002) 5564–5575. DOI:10.1021/jm010057b |
| [17] | Z.L. Wan, Y. Chai, M.L. Liu, H.Y. Guo, Improved synthesis of a gemifloxacin intermediate 4-(aminomethyl) pyrrolidin-3-one-O-methyloxime dihydrochloride. Chin. J. Med. Chem. 19 (2009) 109–111. |
| [18] | L.S. Feng, M.L. Liu, Y.B. Zhang, H.Y. Guo, New way to synthesize DW286-a novel fluoronaphthyridone antibacterial agent. Chem. Res. Chin. Univ. 27 (2011) 981–983. |
| [19] | X. Guo, Synthesis and Antibacterial Activity of Novel Fluoroquinolone Derivatives Containing Substituted Pyrrolidyl/Piperidyl Heterocyclic Moieties at 7-Position, Peking Union Medical College, Beijing, 2011. |
| [20] | C.Y. Hong, Y.K. Kim, J.H. Chang, Novel fluoroquinolone antibacterial agents containing oxime-substituted (aminomethyl) pyrrolidines:synthesis and antibacterial activity of 7-(4-(aminomethyl)-3-(methoxyimino) pyrrolidin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1, 4-dihydro[1, 8] naphthyridine-3-carboxylic acid (LB20304). J. Med. Chem. 40 (1997) 3584–3593. DOI:10.1021/jm970202e |
| [21] | Y. Chai, M.L. Liu, B. Wang, Synthesis and in vitro antibacterial activity of novel fluoroquinolone derivatives containing substituted piperidines. Bioorg. Med. Chem. Lett. 20 (2010) 5195–5198. DOI:10.1016/j.bmcl.2010.07.006 |
| [22] | P. Skehan, R. Storeng, D. Scudiero, New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 82 (1990) 1107–1112. DOI:10.1093/jnci/82.13.1107 |