 2016, Vol.27
2016, Vol.27 


 , Chun-Xiang Hea, Rui-Jing Lib, Hui Gaob, Lin-Ke Geb, Yan-Jie Wangb, Yao Yaob
, Chun-Xiang Hea, Rui-Jing Lib, Hui Gaob, Lin-Ke Geb, Yan-Jie Wangb, Yao Yaob
b. Key Laboratory for Ecological Environment in Coastal Areas (SOA), National Marine Environmental Monitoring Center, Dalian 116023, China
Polychlorinated biphenyls (PCBs), organochlorine pesticides (OCPs), polycyclic aromatic hydrocarbons (PAHs), and hexabromocyclodoecanes (HBCDs), which are listed as persistent toxic substances, exhibit properties similar to those of persistent organic pollutants (POPs). These pollutants are ubiquitous in nature and are found even in remote areas, such as in isolated areas in the polar region. The Antarctic is unique because it serves as an unparalleled natural laboratory for research on problems related to global pollution by persistent toxic substances (PTS) and an ideal site for baseline studies on the behaviors of PTS in the environment. The Antarctic region is mostly covered with snow, ice, and ocean. Although the concentrations of emerging pollutants in the Antarctic region are usually considerably low, these pollutants can be accumulated in sediments and organisms; thus, these pollutants can eventually endanger the ecosystem of the Antarctic region. Their transmission, source apportionment, and degradation can be analyzed when the mechanism of transmission and storage of these emerging pollutants in a multi-medium is fully understood. Therefore, analyzing the typical PTS in Antarctic water is necessary.
Over the past decades, researchers worldwide have developed analytical methods to analyze typical PTS in soil, sediment [1, 2, 3, 4], and biological samples [5, 6, 7, 8]. However, reports on these PTS in water are limited because of the low concentrations of these pollutants in bodies of water. Moreover, pollutants in the polar region have been rarely reported. Most of the published methods for water analysis have focused on specific families, such as pesticides [9, 10, 11, 12], PAHs [13, 14, 15], or HBCDs [16, 17]; only some of these studies could determine various compounds belonging to different families [12, 18]. Furthermore, these studies have mostly focused only on one or two groups of contaminants. Nevertheless, studies have yet to analyze PCBs, OCPs, HBCDs, and PAHs simultaneously. The detection limits of these currently used methods also cannot satisfy the requirements to analyze Antarctic water samples. Sample preparation is of utmost importance in water analysis because these analytes belong to various chemical groups and are present at low concentrations in aquatic media.
To develop a simple and easy-to-use SPE- and gradient elusionbased sample preparation method of various groups of PTS (PCBs, OCPs, HBCDs, and PAHs) in water, we developed an environmentally friendly sample preparation technique that increases the overall sample throughput, eliminates the interference from impurities and target compounds and reduces cost. The factors influencing extraction efficiency to obtain optimum pretreatment conditions, for example sampling volume, extraction solvent, and variety and dosage of elution agent, are investigated.
2. Experimental 2.1. Chemicals and solutionsThe following compounds were acquired from Wellington Laboratories and Cambridge Isotope Laboratories: a mixture standard of the 30 PCBs (8, 18, 28, 44, 52,66, 77, 81,87, 101,105, 110, 114, 118, 123, 126, 128, 138, 153, 1=, 169, 170, 180, 187, 189, 194, 195, 200, 205, and 206); a mixture standard of the 17 OCPs (α- HCH, β-HCH, γ-HCH, δ-HCH, Heptachlor, Aldrin, hextachlor exoxide, γ-chlorcan, α-endosulfan, α-chlorcan, p,p'-DDE, p,p'-DDD, p,p'-DDT, dieldrin, endrin, β-endosulfan, and endrinachyde); a mixture of the 16 PAHs [naphthalene (Nap), acenaphthylene (Ace), acenaphthene, fluorene, phenanthrene (Phe), anthracene, fluoranthene, pyrene, benz(a)anthracene, chrysene, benzo(b)-fluoranthene, benzo(k)- fluoranthene, benzo(a)pyrene, indeno(1,2,3-cd)pyrene, dibenz(a,h)-anthracene, benzo(g,h,i)-perylene]; and a mixture of the 3 HBCDs. The substrate standard used in this study was polychlorinated biphenyl 209 (99%), and the isotope internal standards were Nap-d8, Ace-d10, Phe-d10, Chr-d12, Perylene-d12, Terphenyl-d14, and 13C-γ-HBCD. All of the organic solvents (dichloromethane, n-hexane, methanol, and acetone) used in this study were of HPLC grade (Merck, Darmstadt, Germany). Water was purified using Milli- Qsystem(Molsheim, France). Silica, neutral alumina, and anhydrous sodiumsulfate were purchased from Merck (Darmstadt, Germany). Neutral alumina and anhydrous sodium sulfate were activated for 8 h at 650 ℃ in a muffle furnace, cooled to room temperature, and then stored in a dryer. The silica gel was washed thrice with dichloromethane after the solvent was evaporated to dryness; a glass beaker filled with silica gel was stored in an oven at 170 ℃.
2.2. InstrumentationThe following instruments were used: a gas chromatograph equipped with an electron capture detector (GC-ECD) and an auto injector (Shimadzu, model 2010); a gas chromatograph coupled with a mass spectrometer (GC-MS) and an auto injector (Agilent, model 6890 and MSD 5975B); a liquid chromatograph coupled with a triple-stage quadrupole mass spectrometer TSQ Quantum, equipped with an electrospray ionization source (Thermo scientific) (LC-MS/MS); DB-5 capillary column (30 m × 0.25 mm, 0.25 μm; Agilent); BDS HYPERSIL C18 column (100 mm × 2.1 mm, mm, 2.4 μm, Thermo Scientific); Glass fiber membrane (50 mm, 0.45 μm, Whatman); C18 membrane (47 mm, 3 M Co, USA); organic microporous membrane (0.22 μm, Xinya, Shanghai); solid-phase extraction device (Hengao, Tianjin); semi-automatic solid-phase extraction device (Supelco, USA); and glass column (10 mm × 300 mm).
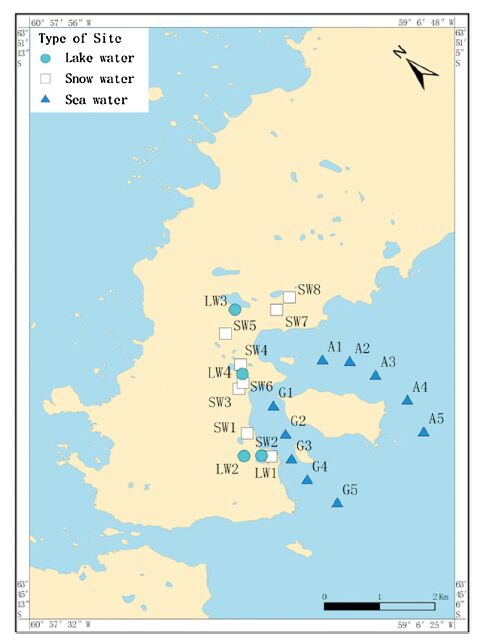
2.3. Sample collectionThe Fildes Peninsula is located southwest of King George Island and receives a marine climate. Fildes Peninsula, where eight scientific stations are found, is rich in biological resources and is the site of intensive scientific investigations in the Antarctic. Sea water, lake water, and snow water samples were collected from 10 (A1-A5 and G1-G5, Fig. 1), 8 (SW1-SW8, Fig. 1), and 4 (LW1-LW4, Fig. 1) sampling sites, respectively. All of these samples were collected from January 2013 to February 2014; from each site, approximately 8 L of water sample was collected in dark glass containers with Teflon cover. Snow samples were collected using stainless steel shovel and aluminum barrel, washed several times with deionized water, and soaked for 24 h with methanol.

|
Download:
|
| Fig. 1.Sampling sites of Fildes Penisula. | |
The water sample (8 L) was measured using a volumetric cylinder, spiked with methanol (5mL L-1), and was shaken well. Each sample was spiked with four different isotopically-labelled internal standards: 50 μL of PCBs and OCPs at 500 ng mL-1 (PCB 209); 50 μL of PAHs at 500 ng mL-1 (Nap-d8, Ace-d10, Phe-d10, Chrd12,Perylene-d12,and Terphenyl-d14); 20 μL of HBCDs at 600 ngmL-1 (13C-γ-HBCD). The calibrated samples were spiked with internal standards and with calibration standards at appropriate concentrations (matrix-matched surrogate standards).
2.4.2. Membrane pretreatment and targets concentrationC18 films were primed with 5 mL dichloromethane-n-hexane (v/v, 1:1), 5 mL methanol, and 5 mL reverse osmosis water. This procedure was performed two times. Most water was effluented and membrane was not exposed to air, which was rendered soluble and activated to avoid interface gap between air and liquid during the entire process. The samples, which were filtered through 0.47 μm glass fiber filters for particle removal, were loaded onto conditioned C18 disks for concentration. Finally, the disks were concentrated to dryness and were wrapped for recovery.
2.4.3. Extraction and gradient elutionThe target compounds were ultrasonically extracted from C18 film using 40 mL dichloromethane-n-hexane (v/v, 1:1). The membrane was soaked for 12 h prior to the ultrasonic extraction. Extraction from each film was performed two times. The two extracts were mixed, concentrated to 3-5 mL by using a rotary evaporator under 40 ℃, and kept for further purification.
Purification was achieved using a multi-layer silica gel column (10 mm -300mm) filled from bottom to top with 2g of anhydrous sodium sulfate, 6.5 g of activated silica gel, 7.5 g of neutral alumina, and 2 g of anhydrous sodium sulfate; the column was then compacted (n-hexane level was higher than that of the filler to avoid bubble formation). The column was pre-washed with n-hexane. The extract was then added and eluted with 50 mL of n-hexane to collect the PCBs and with 60 mL dichloromethane-n-hexane (v/v, 1:1) to collect the mixture of OCPs and HBCDs [19]. The PAHs with a wide range of Kow was collected in both eluants.
2.4.4. Concentration and analysisThe second part of the eluant was divided into two portions: 30 mL of aliquot for GC analysis (OCPs) and 30 mL of aliquot for LC-MS/MS analysis (HBCDs). All of the resulting purified extracts were concentrated to nearly dry using rotary evaporator under 40 ℃. The solutions used in the GC analysis of PCBs and OCPswere reconstituted with 1 and 0.5 mL of n-hexane, respectively. The solution used in LC-MS/MS analysis of HBCDs was reconstituted with 0.5 mL methanol. After the analysis for HBCDs, the solution was further evaporated under a gentle N2 stream, solventexchanged to n-hexane, and then merging the three portions for PAH analysis. The trace or ultra-trace PCBs and OCPs in the water samples were detected using the highly sensitive GC-ECD, a method described byWang [20]. Trace PAHs were detected using GC/MS (EI/SIM), a method described by Ma [21]. Ultra-trace HBCDs were detected using LC-MS/MS, which is an optimized method as described by Wu [22]. All of these methods were established by our own laboratory. The details of these methods were briefly below.
The PCBs and OCPs were analyzed on a GC-ECD (Shimadzu, Model 2010) by using the DB-5 capillary column (30 m × 0.25 mm, 0.25 μm) (Agilent). The injection volume in the splitless mode was 1.0 μL. Helium was used as the carrier gas at a flow rate of 2.0 mL min-1. The gas chromatography oven was programmed as follows: initial temperature was set at 80 ℃ for 2 min, increased to 180 ℃ at 20 ℃ min-1, further increased to 250 ℃ at 4 ℃ min-1 for 2 min, and then increased to 280 ℃ at 30 ℃ min-1 until for 5 min. The injector and detector temperatures were 220 ℃ and 300 ℃, respectively.
The HBCDs were analyzed on an LC-MS/MS coupled with a triple-stage quadrupole mass spectrometer TSQ Quantum, equipped with an electrospray ionization source (Thermo scientific) by using electrospray ionization negative ion and multiple reaction monitoring mode. The injection volume was 20.0 μL. Separations were conducted using BDS HYPERSIL C18 reversedphase column (100 mm × 2.1 mm, 2.4 μm, Thermo Scientific) at a flow rate of 0.2 mL min-1. The program started at an initial composition of 20:20:60 A (methanol)/B (acetonitrile)/C (10 mmol/L CH3COONH4) for 1 min (v/v, hereinafter the same) and was ramped to 70:20:10 A/B/C at 6 min and held for another 7 min, and then changed to 20:20:60 A/B/C at 18 min and held for another 4 min.
The PAHs were analyzed on an Agilent 6890N gas chromatograph coupled with a 5973 I mass spectrometer (MS, Agilent Technologies, Inc., California, USA) by using the negative chemical ionization mode with methane as the ionization gas fitted with a DB-5HT capillary column (0.25 mm i.d. × 30 m × 0.10 μm film thickness, J&W Scientific, Inc., California, USA). The injection volume in the splitless mode was 1.0 μL. Helium was used as the carrier gas at a flow rate of 1.0 mL min-1. The gas chromatography oven was programmed as follows: initial 50 ℃, increased to 220 ℃ at 4 ℃ min-1 and held for 3 min, and then further increased to 300 ℃ at 5 ℃ min-1 and held for 9 min. The MS transfer line was held at 290 ℃. The temperature of the ion source and the quadrupole were 230 ℃ and 150 ℃, respectively. The instrument was operated in selected ion monitoring mode.
2.5. QuantificationThe compounds were quantified using the matrix-matched surrogate calibration method. All of the samples were spiked with a suitable internal standard prior to SPE; the calibration samples were also spiked with a mixture of standards to obtain then final increasing concentration. The calibration curve was generated by plotting the ratio of the analyte peak area to the internal standard peak area against the concentration. This method reduced the matrix, recovery, and instrumental precision effects.
3. Results and discussion 3.1. Preconcentration optimizationOwing to the harsh conditions in the region, the Antarctic is least affected by anthropogenic activities compared with other parts of the earth. These harsh conditions limit the transport of large volumes of water samples for analysis. Therefore, analyte preconcentration is necessary. Compared with other preconcentration methods, SPE is preferable because of its advantages, such as wide range of applications, ease of use and enrichment, low solvent consumption, highly efficient enrichment, low requirement for manpower and materials, and avoidance of emulsification; SPE has also been recommended for EPA. The surface area of SPE disks is larger by 10 times than that of SPE cartridges. Moreover, high-velocity SPE disks are difficult to block. Therefore, the SPE membrane was selected in enriching the analytes of the water samples obtained from the Antarctic. Finally, C18 film was selected to enrich targets which could solve the transportation of large volume water samples and solve the simultaneous enrichment of the multi class compounds.
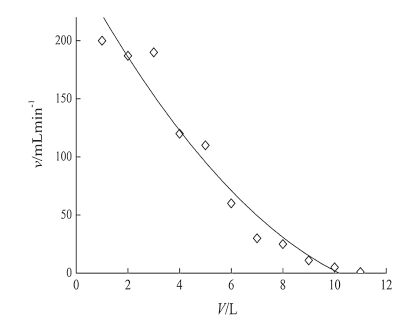
3.2. Volume optimizationThe optimal sampling volume was determined as follows: (1) the minimum sampling volume was 8 L based on the limit of detection (LOD) and the recovery of HBCDs (the lowest level); and (2) basing on the flow curve, we used GF/F glass fiber (47 mm × 0.47 μm) to filter impurities of sizes larger than 0.47 μm × 47 μm) and then the water was loaded onto the C18 membrane. As the volume increases, micropores of the C18 membrane are plugged, thereby reducing the flow rate. Fig. 2 shows the velocity curve.

|
Download:
|
| Fig. 2.The velocity curve of target compounds enrichment. | |
Basing from Fig. 2,we may conclude that the flow rate is negatively correlated with sample volume; the flow rate was close to the minimum value at a sampling volume of 10 L, whereas the flow rate is 25 mL min-1 at 8 L, which is consistent with EPA recommendation. Thus, the sampling volume was 8 L.
3.3. Extraction agent optimizationThe lg Kow values of PCBs, OCPs, HBCDs, and PAHs were 4.46- 8.18, 3.3-6.9, 5.4-5.8, and 3.3-6.6, respectively [23]. The lg Kow value represents the hydrophobicity and lipid solubility of organic compounds; high lg Kow value indicates high dissolution of a compound in non-polar medium (organic phase). The substances with lg Kow of >4 are strongly hydrophobic. The ascending sequence of polarity of the investigated compounds is as follows: PCBs < OCPs, HBCDs, and PAHs. Moreover, the vapor pressures of PCBs, OCPs, HBCDs, PAHs were 1.1-6.3 × 10-6, 0.2-2.8 × 10-7, 6.3 × 10-5, and 1.45 × 10-1-6.2 × 10-14, respectively [23]. This result indicated that PCBs, OCPs, HBCDs, and PAHs are semivolatile (10-4-10-11), non-polar or weakly polar hydrophobic organic compounds; in addition, they are soluble in hexane, dichloromethane, acetone, and other organic solvents exhibiting a similar polarity.
n-Hexane, dichloromethane, ethyl acetate, and their binary mixtures (hexane-acetone and hexane-dichloromethane) are commonly used worldwide and they all produce satisfactory results. The results in this study are shown in Table 1.
|
|
Table 1 Comparison of analytical methods used for determination of target compounds in water. |

The extraction efficiency was selected to optimize the extraction conditions. Basing on the theory of similarity and intermiscibility and from the recoveries presented above, the polarity of extraction solvent should be nearly similar or slightly stronger than that of the target compounds, also it should be ensure that the solubility of impurities should be as small as possible. Ethyl acetate (lg Kow is 0.73), whose viscosity and boiling point are high, is therefore not conducive for target extraction. In general, the binary-solvent mixtures were used to improve the penetration of the matrix and to improve the enrichment of multiclass contaminants in complex matrices. On the basis of EPA 505, 508, 525, and the extraction results above, we selected hexane- dichloromethane as the extraction solvent at various ratios (v/v, 1:1,1:2,2:1,2:3, and 3:1). The highest recovery was achieved with dichloromethane-hexane (v/v, 1:1).
3.4. Purification and separationEPA sw-846 series sample purification and EPA 3630 silica gel purification revealed that silica gel column provides good chemical stability, high mechanical strength, and wide range of applications. As target compoundswere non-polar or weak-polar, silica gel column was chosen to purify the targets. Impurities were passed through the silica gel column and then adsorbed on the surface of the active agent. On the basis of different Kow of analytes, we found that the first fraction containing PCBs and part OCPs was eluted with non--polar hexane; the second fraction containing OCPs and HBCDs was eluted with dichloromethane- hexane (v/v, 1:1) because of the polarity of the analytes increased; PAHs were maintained in binary-solvent mixtures. Furthermore, fat, wax, pigments, and other impurities were retained in a column, and the objectives of separation and purification were attained.
After homogeneous mixing was performed, the second part of the eluant was divided equally into two portions, one portion was used to analyze OCPs and the other portion was utilized to analyze HBCDs. The eluant was divided into two aliquots to reduce the loss caused by solvent replacement (from GC to LC-MS/MS). Furthermore, accurate qualitative and quantitative characterization of the substances distributed in various eluants (such as DDE and Aldrin; endosulfan and alpha chlordane) through gradient elution and mutual interference is difficult to achieve because different groups in a category exhibit similar properties.
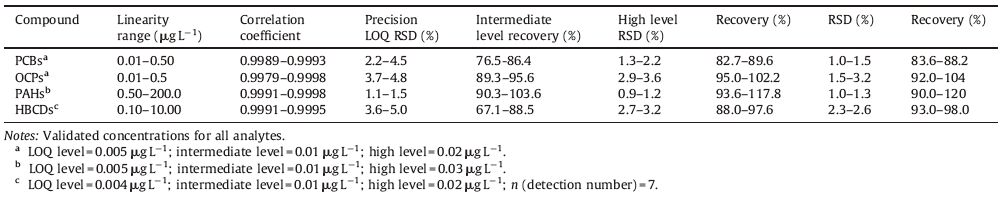
3.5. Analytical method validationTo validate the developed method to quantify water pollutants, we assessed the linearity, selectivity, LOD, quantification, and precision of the experimental procedure. Linearity and recovery were evaluated by spiking the deionized water with varying amounts of the compounds to obtain the final concentrations. The results are shown in Table 2. The LOD and the limit of quantification are the lowest concentrations that produce a chromatographic peak at a signal-to-noise ratio of 3 and 10, respectively. A field blank, a procedural blank, and a reagent blank were analyzed every eight samples to verify field, laboratory, and reagent contaminations; all of the blank concentrations were below LOD levels of the target compounds.
|
|
Table 2 Validation results. |
{kind=link}
{kind=link}
The method detection limits (MDLs) were derived from the instrumental detection limits. The MDL ranges of PCBs, OCPs, HBCDs, and PAHs were 0.002-0.018, 0.002-0.018, and 0.004- 0.02 ng L-1, and 0.02-0.20 ng L-1, respectively.
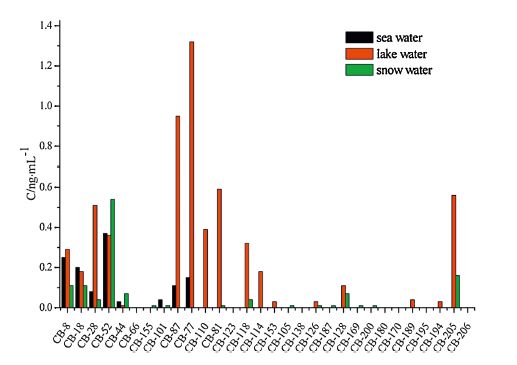
3.6. Analysis of real samplesThe method was successfully applied in analyzing 10 sea water samples, 4 lake water samples, and 8 snow water samples collected from the Fildes Peninsula in the Antarctic. Results are shown in Figs. 3-6.

|
Download:
|
| Fig. 3.The mean concentration of PCBs. | |
{kind=link}

|
Download:
|
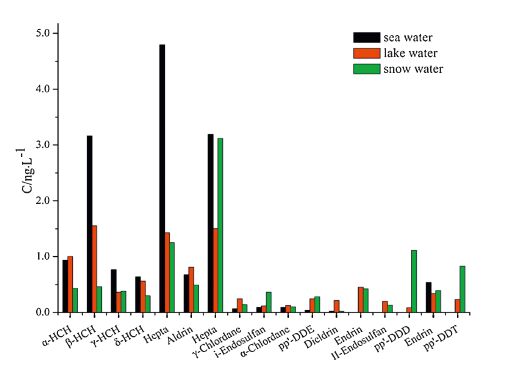
| Fig. 4.The mean concentration of OCPs. | |
{kind=link}

|
Download:
|
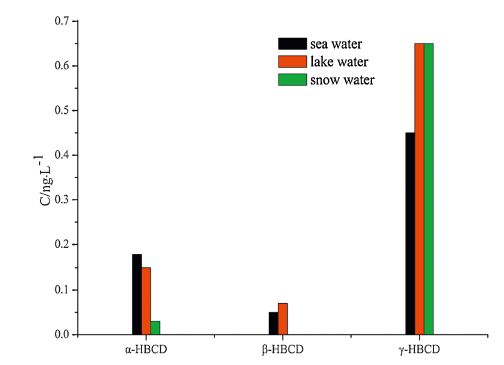
| Fig. 5.The mean concentration of HBCDs. | |
{kind=link}

|
Download:
|
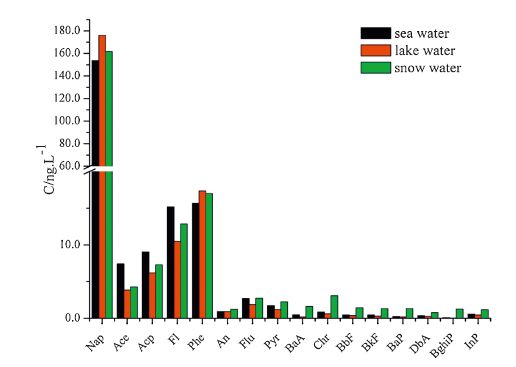
| Fig. 6.The mean concentration of PAHs. | |
{kind=link}
Analysis revealed the concentration of the compounds in the samples to be as follows: PCBs: 0.81-3.16 ng L-1 in seawater, 0.88- 2.89 ng L-1 in lake water, and 0.85-2.33 ng L-1 in snow water; OCPs: 5.69-21.46 ng L-1 in seawater, 8.06-11.22 ng L-1 in lake water, and 2.69-22.81 ng L-1 in snow water; HBCDs: 0.27- 1.58 ng L-1 in seawater, 0.48-1.54 ng L-1 in lake water, and 0.13-1.16 ng L-1 in snow water; PAHs: 117.12-327.69 ng L-1 in seawater, 145.56-339.44 ng L-1 in lake water, and 89.19- 324.59 ng L-1 in snow water. The concentrations of the target compounds were relatively low in the Antarctic. PCBs containing fewer chlorine atoms (low molecular weight) is easily transferred into the cooler region, whereas PCBs containing more chlorine atoms (high molecular weight) tends to settle near the source. The amount of β-HCH was higher than α,γ,δ-HCH possibly because β- HCH exhibits the highest symmetry and a stable physicochemical properties. Compared with the other targets, higher concentration of PAHs was detected; PAHs with 2-3 rings were mostly observed (92.37%-97.54%) possibly because low-molecular-weight PAHs exhibit high mobility and thus easily move from the lower latitude to higher latitude through the global distillation effect. Lowmolecular- weight PAHs (2-3 rings) mainly cause oil pollution, which is in compliance with scientific investigations commonly used diesel for power. Highly soluble γ-HBCD is the dominant compound in all of the water samples.
4. ConclusionAn analytical method was developed and validated to perform the simultaneous concentration and selective separation of 66 (ultra) trace multiclass environmental contaminants in Antarctic waters. Compared with the reported methods to analyze PCBs, OCPs, HBCDs, and PAHs, the proposed method provides advantages in terms of recovery, selectivity, sensitivity, and LOD. This new method can be applied to concentrate large volume water samples and separate typical, multi class PTS in remote regions. The method could be used as an experimental basis of further analysis of migration regularity and environmental fate of the typical PTS.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (No. 21377032), the Chinese Polar Environment Comprehensive Investigation and Assessment Program (Nos. 2014-02-01,2014-03-04, 2014-04-01,2014-04-03), the Marine Public Welfare Scientific Research Projects (No.201105013) and the Foundation of Polar Science Key Laboratory, SOA, China (No. KP201208).
| [1] | M.A.E. Abdallah, D. Drage, S. Harrad, A one-step extraction/clean-up method for determination of PCBs, PBDEs and HBCDs in environmental solid matrices, Environ. Sci. Pro. (Ⅰ)mpacts 15(2013) 2279-2287. |
| [2] | L. Wolska, Miniaturised analytical procedure of determining polycyclic aromatic hydrocarbons and polychlorinated biphenyls in bottom sediments, J. Chromatogr. A 959(2002) 173-180. |
| [3] | A. Mahmood, R.N. Malik, J. Li, G. Zhang, Levels, distribution pattern and ecological risk assessment of organochlorines pesticides (OCPs) in water and sediments from two tributaries of the Chenab River, Pakistan, Ecotoxicology 23(2014) 1713-1721. |
| [4] | R. Sarria-Villa,W. Ocampo-Duque, M. Páez, M. Schuhmacher, Presence of PAHs in water and sediments of the Colombian Cauca River during heavy rain episodes, and implications for risk assessment, Sci. Total Environ. (2015), doi:10.1016. |
| [5] | M.(Ⅰ).H. Helaleh, A. Al-Rashdan, A. (Ⅰ)btisam, Simultaneous analysis of organochlorinated pesticides (OCPs) and polychlorinated biphenyls (PCBs) from marine samples using automated pressurized liquid extraction (PLE) and Power PrepTM clean-up, Talanta 94(2012) 44-49. |
| [6] | G.B. Kim, H.M. Stapleton, PBDEs, methoxylated PBDEs and HBCDs in Japanese common squid (Todarodes pacificus) from Korean offshore waters, Mar. Pollut. Bull. 60(2010) 935-940. |
| [7] | G.L. Mihalca, O. Tiţa, M. Tiţa, A. Mihalca, Polycyclic aromatic hydrocarbons (PAHs) in smoked fish from three smoke-houses in Braşov County, J. Agroaliment. Proc. Technol. 17(2011) 392-397. |
| [8] | M.H. Son, J. Kim, E.S. Shin, S.H. Seo, Y.S. Chang, Diastereoisomer-and speciesspecific distribution of hexabromocyclododecane (HBCD) in fish and marine invertebrates, J. Hazard. Mater. 300(2015) 114-120. |
| [9] | Y.M. Sun, Y.F. Wang, F. Liu, et al., Determining semi-volatile organic substances of groundwater through liquid-liquid extraction-C18 membrane disk extraction-GS/MS, J. Chin. Mass. Spectr. Soc. 27(2006) 140-147. |
| [10] | B.H. Jin, F. Xiao, B. Chen, P.J. Chen, L.Q. Xie, Determination of 27 poison compounds in bottled drinking water by combining C18 solid-phase membrane extraction with GC-MS technique, Chin. J. Anal. Lab. 28(2009) 99-102. |
| [11] | T. Trtić-Petrović, J. Dordević, N. Dujaković, et al., Determination of selected pesticides in environmental water by employing liquid-phase microextraction and liquid chromatography-tandem mass spectrometry, Anal. Bioanal. Chem. 397(2010) 2233-2243. |
| [12] | N. Dujaković, S. Grujić, M. Radišić, T. Vasiljević, M. Laušević, Determination of pesticides in surface and ground waters by liquid chromatography-electrospray-tandem mass spectrometry, Anal. Chim. Acta 678(2010) 63-72. |
| [13] | W. Kanchanamayoon, N. Tatrahun, Determination of polycyclic aromatic hydrocarbons in water samples by solid phase extraction and gas chromatography, World J. Chem. 3(2008) 51-54. |
| [14] | (Ⅰ). Windal, L. Boxus, V. Hanot, Validation of the analysis of the 15+1 Europeanpriority polycyclic aromatic hydrocarbons by donnor-acceptor complex chromatography and high-performance liquid chromatography-ultraviolet/fluorescence detection, J. Chromatogr. A 1212(2008) 16-22. |
| [15] | Y.L. Chao, H. Zhou, C.Q. Yu, et al., Determination of PAHs in snow from Hailuogou by C18 membrane extraction chromatography spectrometry, Liaoning Chem. (Ⅰ)nd. 43(2014) 498-502. |
| [16] | M. (Ⅰ)chihara, A. Yamamoto, K.(Ⅰ). Takakura, N. Kakutani, M. Sudo, Distribution and pollutant load of hexabromocyclododecane (HBCD) in sewage treatment plants and water from Japanese Rivers, Chemosphere 110(2014) 78-84. |
| [17] | Y. Liu, K. Hu, X.P. Ye, Progress in research of analysis on the hexabromocyclododecane, Guangdong Chem. (Ⅰ)nd. 39(2012) 296-297. |
| [18] | J. Sánchez-Avila, M. Fernandez-Sanjuan, J. Vicente, S. Lacorte, Development of a multi-residue method for the determination of organic micropollutants in water, sediment and mussels using gas chromatography-tandem mass spectrometry, J. Chromatogr. A 1218(2011) 6799-6811. |
| [19] | K. Kalachova, J. Pulkrabova, T. Cajka, et al., Gas chromatography-triple quadrupole tandem mass spectrometry:a powerful tool for the (ultra) trace analysis of multiclass environmental contaminants in fish and fish feed, Anal. Bioanal. Chem. 405(2013) 7803-7815. |
| [20] | Y.J. Wang, L. Li, X.D. Ma, et al., Determination of organochlorine pesticides and PCBs in sea water by silica gel column separation and gas chromatography, Environ. Monitor. China 25(2009) 39-41. |
| [21] | X.D. Ma, Z.W. Yao, Z. Wang, et al., Distribution and environmental behavior of PAHs in different matrixes on the Fildes Peninsula, Antarctica, Advan. Polar Sci. 26(2014) 285-291. |
| [22] | X. Wu, G.R. Zu, H. Gao, et al., Distribution characteristic and bioaccumulation of hexabromocyclododecanes (HBCDs) in multimedia environment in the coast of northern Yellow Sea, J. Environ. Chem. 33(2014) 142-147. |
| [23] | J.P. Chen, Q. Fu, Monitoring Technology of Persistent Organic Pollutants in Water, Chemical (Ⅰ)ndustry Press, Beijing, 2015. |
| [24] | M.J. Deng, Y.N. Luo, C.B. Deng, Determination of 19 polychlorinated biphenyls in water by gas chromatography-mass spectrometry with solid phase extraction, Chin. J. Spectr. Lab. 29(2012) 947-950. |
| [25] | S.D. Yu, Determination of polycyclic aromatic hydrocarbons, PCBs, and phthalats by SPE disk extraction-GC/MS with selected ion monitoring mode, Anal. TestingTechnol. (Ⅰ)nstrum. 12(2006) 161-165. |
| [26] | M. Vecchiato, E. Argiriadis, S. Zambon, et al., Persistent organic pollutants (POPs) in Antarctica:occurrence in continental and coastal surface snow, Microchem. J. 119(2015) 75-82. |
| [27] | R. Fuoco, S. Giannarelli, M. Onor, et al., A snow/firn four-century record of polycyclic aromatic hydrocarbons (PAHs) and polychlorobiphenyls (PCBs) at Talos Dome (Antarctica), Microchem. J. 105(2012) 133-141. |
| [28] | E. Martinez, M. Gros, S. Lacorte, D. Barceló, Simplified procedures for the analysis of polycyclic aromatic hydrocarbons in water, sediments and mussels, J. Chromatogr. A 1047(2004) 181-188. |
| [29] | G.S. Na, C.Y. Liu, Z. Wang, et al., Distribution and characteristic of PAHs in snow of Fildes Peninsula, J. Environ. Sci. 23(2011) 1445-1451. |
| [30] | Y.F. Wang, F. Liu, Y.M. Sun, F. Shi, Enrichment of trace amount of semi-volatile organic pollutants from large volumes of ground water by extraction with solidphase membrane, Phys. Testing Chem. Anal. 44(2008) 127-130. |
| [31] | S. Suzuki, A. Hasegawa, Determination of hexabromocyclododecane diastereoisomers and tetrabromobisphenol A in water and sediment by liquid chromatography/mass spectrometry, Anal. Sci. 22(2006) 469-474. |
| [32] | L.X. Wang, S.H. Qi, X.G. Wu, et al., Spatial and temporal variations of organochlorine pesticides (OCPs) in water and sediments from Honghu Lake, China, J. Geochem. Explor. 132(2013) 181-187. |
| [33] | D. Yang, S.H. Qi, J.Q. Zhang, et al., Organochlorine pesticides in soil, water and sediment along the Jinjiang River mainstream to Quanzhou Bay, Southeast China, Ecotoxicol. Eniron. Saf. 89(2013) 59-65. |
| [34] | Y. Hu, S.H. Qi, J.P. Zhang, et al., Assessment of organochlorine pesticides contamination in underground rivers in Chongqing, Southwest China, J. Geochem. Explor. 111(2011) 47-55. |
| [35] | Y. Yu, Y.X. Li, Z.Y. Shen, et al., Occurrence and possible sources of organochlorine pesticides (OCPs) and polychlorinated biphenyls (PCBs) along the Chao River, China, Chemosphere 114(2014) 136-143. |