 2016, Vol.27
2016, Vol.27 



b School of Pharmaceutical Engineering, Shenyang Pharmaceutical University, Shenyang 110000, China;
c The Graduate School at Shenzhen, Tsinghua University, Shenzhen 518000, China
Angiogenesis,the process by which new blood vessels grow from a body’s vasculature,is fundamental to physiological processes of reproduction and wound healing. Disturbances in this process are associated with pathological conditions like rheumatoid arthritis,age-related macular degeneration,diabetic retinopathy,and are highly related to tumor progression and metastasis [1, 2]. It has been confirmed that blocking angiogenesis is an efficient and prospective approach for cancer therapy. VEGFRs (vascular endothelial growth factor receptors) are key regulatory and signaling molecules involved in angiogenesis and consist of three subtypes: VEGFR-1,VEGFR-2,and VEGFR-3 [3]. Overexpression of VEGFRs serves as a potential target for anticancer agents. In other words,a drug can be designed based on the overexpressed genetic marker. Currently an extensive array of VEGFR inhibitors is entering clinics and/or achieving approval from FDA,such as bevacizumab,sorafenib,sunitinib,pazopanib and vandetanib, while many are still in preclinical development [4, 5].
The phosphatidylinositol 3-kinases (PI3Ks) are members of a unique group of intracellular lipid kinases [6]. The PI3K family is involved in numerous cellular functions including proliferation, adhesion,migtation,invasion,metabolism and survival [7]. Frequent occurrences of aberrant signaling mediated by PI3Ks in human cancers have made them attractive targets for the design of small molecule inhibitors.
Although antiangiogenesis has shown to be a promising strategy for cancer therapy,VEGFR inhibitors often encounter resistance to novel therapeutic agents or chemotherapeutics after a period of treatment. One important explanation is that many other pathways are activated during antiangiogenic treatment to counteract the therapeutic efficacy [8]. The PI3K/Akt signaling pathway is one that has proven to be a bypass or compensatory pathway and can become overactive in the presence of cancers or certain agents [9, 10]. PI3Ks upregulate angiogenic cytokines due to tumor hypoxia,or oncogene stimulation,and alter endothelial cell responses to them. These cytokines signal through the receptors VEGFR,FGFR,and Tie-2 to potentiate cell proliferation,migration, differentiation into tubules,and "invasion" of these capillary sprouts into extracellular matrix [11]. Therefore,blocking activation of the PI3K/Akt pathway during antiangiogenesis therapy could reduce tumor progression.
However,VEGFRs and PI3Ks are from different kinases families so it is a challenge to effectively design dual inhibitors of VEGFRs and PI3Ks. As of now,there are no reports on an agent that targets both enzymes. Our investigation was induced by the lack of such an agent and the possibility of discovering VEGFR and PI3K dual inhibitors with novel scaffold. With this purpose,we searched the Ambinter and Chemspider libraries by the combinatory approach of SVM (support vector machine) and docking and were able to identify a one hit compound 5a containing imidazo[2, 1, b]thiazole scaffold. Based on this hit compound,a series of 2-aminothiazol-4- acetamide and 2-aminothiazol-4-carboxamide derivatives was designed and synthesized. All compounds were evaluated for their in vitro cytotoxicity against human HepG2 and MDA-MB-231 cell lines. Kinase inhibition and molecular docking were also studied. The results showed that compounds 6i and 6j containing 2- ureidothiazol scaffold have good PI3K and moderate VEGRF inhibitory activity along with potency against both MDA-MB- 231 and HepG2 cell lines.
2. Experimental 2.1. General1H NMR,13C NMR spectra were determined on Bruker ARX-400, 400 Hz spectrometers with tetramethylsilane (TMS) as the internal standard and DMSO-d6,CDCl3 as the solvent (Chemical shifts in ppm). Splitting patterns were designated as follows: s: singlet; d: doublet; t: triplet; m: multiplet. Mass spectra were carried out using a Waters Micromass Q-TOF Premier Mass Spectrometer. Melting points were determined in open glass capillaries with a SGW X-4 digital apparatus and were uncorrected. Follow-up of the reactions and checking the homogeneity of the compounds were made by TLC on silica gel-protected glass plates and the spots were detected by exposure to UV-lamp at λ254 and λ365. Unless otherwise noted,all solvents and reagents were commercially available and used without further purification.
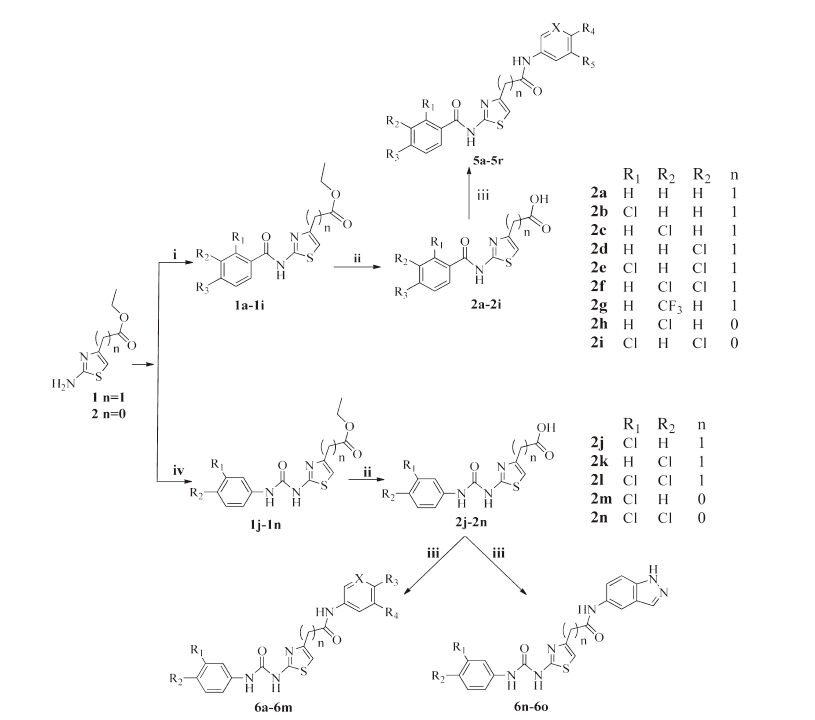
2.2. General procedure for preparation of compounds 2a-2nTo a solution of compound ethyl 2-aminothiazole-4-acetate (1) (10 mmol) or ethyl 2-aminothiazole-4-carboxylate (2) (10 mmol) in anhydrous THF (10 mL),NMM (15 mmol) and substituted benzoyl chlorides (11 mmol) or substituted isocyanatobenzenes (11 mmol) were added with stirring at r.t. for 12 h. The solution was cautiously basified with 15% NH4OH to pH 7,then poured into CH2Cl2,separated,washed and concentrated to result in four kinds of esters 1a-1n,respectively. The crude product 1a-1n was dissolved in EtOH-H2O-NaOH (700 mL:300 mL:60 g) and refluxed for another 0.5 h,then acidified to pH 3-4 with concentrated HCl to afford white solid precipitation. After filtration,wash with water and dry,the desired compounds 2a-2n were obtained.
2.3. General procedure for preparation of compounds 3a-3b,4a-4bA mixture of 2-chloro-5-nitropyridine (3) (50 mmol),morpholine (100 mmol),and K2CO3 (100 mmol) in THF (50 mL) were stirred at 80 ℃ for 4 h. And then the mixture was concentrated to 20 mL and poured into water (100 mL),the yellow solid precipitation formed. After filtration,wash with purified water and dry,the desired compound 3a was obtained. 3b Was got with the same method. The mixture of 3a-3b (20 mmol) and Pd/C (20%, 500 mg) in methanol was hydrogenated at atmosphere at r.t. for 12 h,followed by filtration and concentration to afford compounds 4a-4b.
2.4. General procedure for preparation of compound 4c5-Amino-2-hydroxybenzoic acid (4) (100 mmol),methanol (80 mL) and H2SO4 (15 mL) were stirred at 80 ℃ for 24 h,then cold to r.t. and pale yellow solid precipitation formed. After filtration,the crude product was dissolved in 200 mL ethyl acetate and 100 mL H2O. The resulting solution was cautiously basified with 15% NH4OH to pH 8-9,then separated,washed and concentrated to get 4c.
2.5. General procedure for preparation of compounds 5a-5r,6a-6oA solution of the 2a (1 mmol) and 4a (1 mmol),EDCI (1 mmol), HOBt (1 mmol),and DIPEA (3 mmol) in anhydrous THF (10 mL) was stirred for 24 h. The reaction was quenched with 1 mol/L NaOH (20 mL) and extracted with ethyl acetate (3 × 20 mL),the organic layer was washed with 1 mol/L HCl (3 × 20 mL),water (20 mL),dried with Na2SO4 and evaporated to give compound 5a as a white solid. Other title compounds 5b-5r,6a-6o were synthesized as the same procedure. Procedures for preparation of the compounds not mentioned here could be find in Supplementary data. The characterization data for the synthesized compounds are deposited in Supporting information.
2.6. Molecular docking methodologyThe molecular docking of the representative compound 6i with kinases was carried out using Discovery Studio.3.1/CDOCKER protocol (Accelrys Software Inc.). The protein crystallographic structure,VEGFR (PDB entry 2QU5) and PI3K (PDB entry 3L54) were downloaded from the Protein Data Bank (PDB). The general procedure is as followed: (1) ligand and receptor preparation,(2) protocol generation,(3) docking and (4) analysis of the results.
3. Results and discussion 3.1. High-throughput virtual screeningVirtual screening against Ambinter and in-house libraries was conducted using the similar methods and computational procedures as those described in a recently published paper [12, 13, 14]. Initially,SVM models of VEGFR and PI3K inhibitors were used to screen the compounds and received many SVM virtual hits. The hits were evaluated by Lipinsky’s rule of five,and compounds that passed Lipinsky’s rule were selected for further screening via molecular docking. Finally,compound 5a (Ambinter ID 15893411) was identified as a virtual multi-target VEGFR and PI3K inhibitor. This compound and the other derivatives of 5a were synthesized. The kinase assay result suggested that 2-aminothiazole is a potential scaffold and can be modified to get novel compounds with better antitumor activity.
3.2. ChemistryThe hit compound 5a and target compounds 5b-5r,6a-6o collected in Table 1 were prepared as shown in Scheme 1. Ethyl 2- aminothiazole-4-acetate (1) and ethyl 2-aminothiazole-4-carboxylate (2) were reacted with substituted benzoyl chlorides and substituted isocyanatobenzenes resulting in four different esters: 1a-1n. Compounds 1a-1n were directly refluxed in EtOH-H2O- NaOH and acidified to get 2a-2b [15]. Compounds 3a-3b were obtained from 2-chloro-5-nitropyridine (3) and other amines as shown in Scheme 2 [16]. The nitro-group on compounds 3a-3b was reduced to an amino group by hydrogen using Pd/C as catalyst at atmospheric pressure and room temperature to get compounds 4a-4b as shown in Scheme 2. 5-Amino-2-hydroxybenzoic acid (4) was esterified with methanol and catalyzed by H2SO4 to get compound 4c. The target compounds were prepared via coupling 2a-2n with 4a-4c and other commercially available amines using DIPEA,HOBt,EDCI [17]. If necessary,silica-gel column chromatography was used with dichloromethane-methanol system as eluant.
|
|
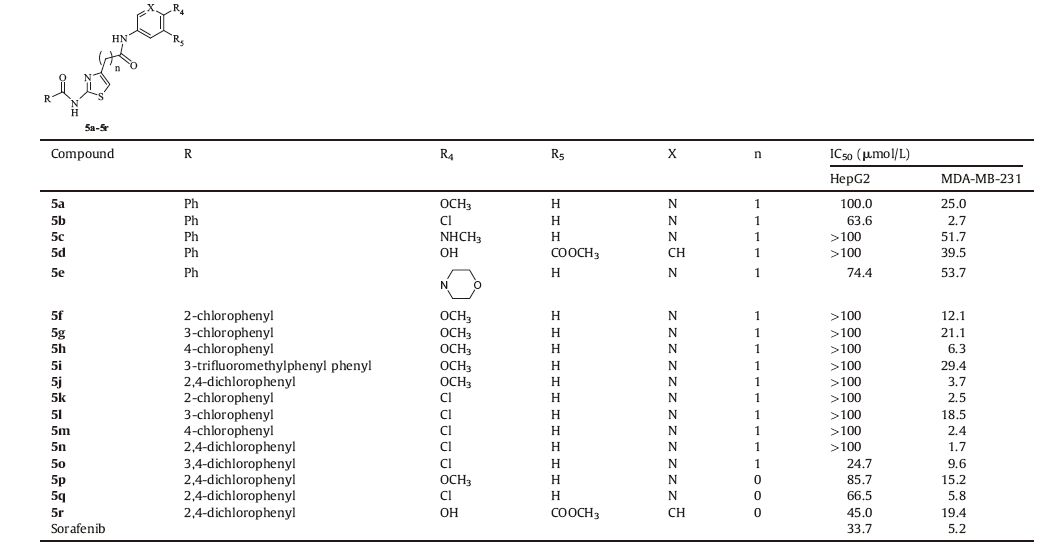
Table 1 The 2-benzamidothiazole-4-amide derivatives and IC50 values against the liver cancer cell and human breast cancer cell line of compounds 5a-5r. |

|
Download:
|
| Scheme 1.Synthesis of compounds 2a-2n,5a-5r and 6a-6o. Reagents and conditions: (i) THF,substituted benzoyl chlorides,12 h,r.t.; (ii) EtOH-H2O-NaOH (1.5 mol/L), reflux,0.5 h,then HCl (2 mol/L); (iii) amines,DIPEA,HOBt,EDCI,THF,12 h,r.t.; (iv) THF,substituted isocyanatobenzenes,12 h,r.t. | |

|
Download:
|
| Scheme 2.Synthesis of compounds 4a-4c. Reagents and conditions: (i) morpholine or methylamine hydrochloride THF,K2CO3,reflux,4 h; (ii) H2,Pd/C,r.t.,12 h; (iii) methanol,H2SO4,reflux,24 h. | |
As shown in Table 1,the hit compound 5a and other 2- benzamidothiazol-4-amide derivatives 5b-5r were evaluated against HepG2 and MDA-MB-231 cell lines with Sorafenib as the positive control by MTT assay. The cells were treated with compounds in the range of 0.1-100 μmol/L for 48 h.
The results showed that these compounds have selective inhibition against the MDA-MB-231 cell line compared to the HepG2 cell line. Five of the compounds that have IC50 values ranging from 1.7 μmol/L to 3.7 μmol/L were more potent than Sorafenib (IC50 5.2 μmol/L). Compounds 5a,5f-5j,5p possess a methoxy group while compounds 5b,5k-5o,5q possess a chloro group on position 2 of the pyridine ring. The chloro compounds were more potent than the corresponding methoxyl derivatives. The activity of 5f-5j for the MDA-MB-231 cell line gradually increased in conjunction with substitutions by 3-chloro,2-chloro, 4-chloro,and 2,4-chloro on the benzene ring and this sequence was consistent with the activity of compounds 5k-5n. However, compound 5o with 3,4-dichloro substituted on benzene ring can more so increase the activity against HepG2 and decrease the activity for MDA-MB-231 compared to compound 5n,which was the 2,4-dichloro substituted on the benzene ring. The activity of compound 5e showed that pyridine substituted by a bulk group increased the activity for HepG2 and decreased the activity for MDA-MB-231 when contrasted to hit compound 5a. After removal of the methane group to yield derivatives of compound 2,the activity for HepG2 increased while the activity for MDA-MB-231 remained unaltered,as suggested by 5p vs 5j,5q vs 5n. In an attempt to replace the pyridine ring with to get compound 5r,the activity stayed moderate against the two cell lines. The above observation indicated that a pyridine ring on position 2 substituted by a chloro group was crucial for antitumor activity.
Based on 2-benzamidothiazole derivatives,a series of 2-(3- phenyl)ureidothiazole derivatives containing a urea group was synthesized as listed in Table 2. Overall,the cyototoxicity activity against HepG2 greatly increased while the activity for MDA-MB- 231 remained constant. Compounds 6f and 6g-6o have IC50 values ranging from 6.4 μmol/L to 20.8 μmol/L for HepG2,and 6d-6o have IC50 values ranging from 1.6 μmol/L to 4.6 μmol/L for MDAMB- 231. These compounds were more potent than Sorafenib (IC50 33.7 μmol/L and 5.2 μmol/L,respectively).
|
|
Table 2 The 2-(3-phenyl) ureidothiazole-4-amide derivatives and IC50 values against the liver cancer cell and human breast cancer cell line of compounds 6a-6o. |
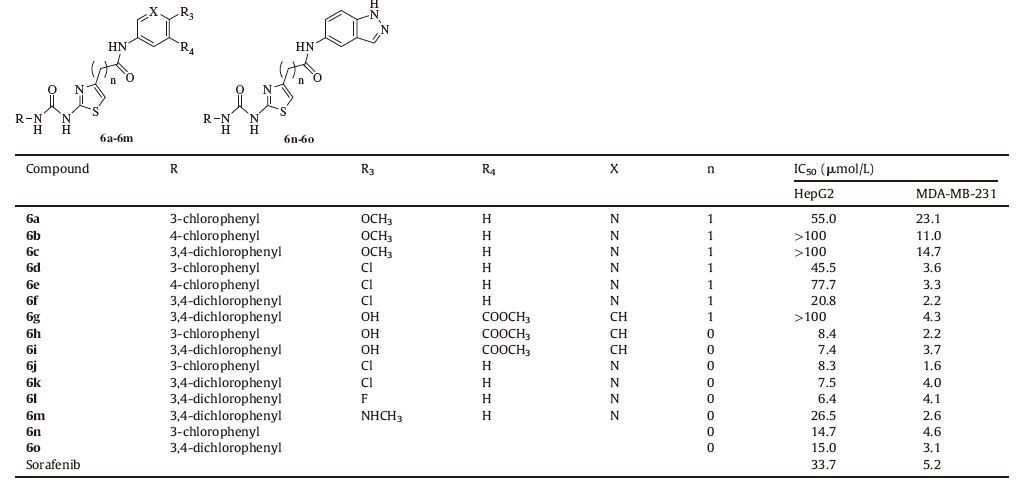
SARs suggested that substitution by 3-chloro on the benzene ring more reactive than the substitution of 4-chloro for HepG2,6a vs 6b,and 6d vs 6e. Contrarily,the results for the MDA-MB-231 cell line were the reverse of the latter. 3,4-Dichloro substituted on benzene ring increased the activity in both HepG2 and MDA-MB- 231. Similar results were produced for the 2-benzamidothiazole derivatives in which the chloro group was better than the methoxy group on position 2 of the pyridine ring.
In order to increase the rigidity of compounds and shorten the length of the chain,methane was removed in order to synthesize a series of 2-(3-phenyl)ureidothiazole-4-formamide derivatives 6h- 6o. The best results were obtained with compounds 6h-6l. This may be attributed to hydrogen bond formation at the receptor site. Basis on compounds 5d,5r,6g,compounds 6h,6i were synthesized and exhibited good activity in the two cell lines. Compound 6l was produced by exchanging the chloro group with a fluoro group on 6k. The activity had no significant increase. We tried changing the pyridine ring to indazole and received compounds 6n and 6o, which resulted in decreased activity against HepG2 and the same activity for MDA-MB-231.
By observing anticancer activity from the data in Table 1 and Table 2,it was concluded that compounds 5a-5r possess an amide linker while compounds 6a-6o have a urea group linker. The best results were obtained with compounds 6h-6l,which exhibited high potency against HepG2 and MDA-MB-231 cell lines with a urea linker. It was decided that the urea and 4-formamide are optimum for this series of compounds against HepG2 and MDAMB- 231 cell lines.
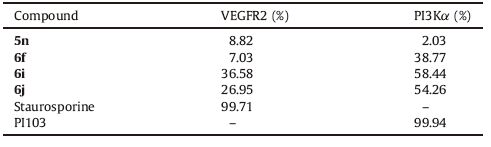
3.3.2. Kinase inhibitionCompounds 5n,6f,6i and 6j were selected for further evaluation in VEGFR2 and PI3Kα kinases inhibition assays at the concentration of 20 μmol/L with Staurosporine and PI103 as controls,respectively. As the kinase assay results reveal in Table 3 compound 5n showed no obvious kinase inhibition against both VEGFR2 and PI3K kinases. Compound 6f showed selective kinases inhibition with lower for VEGFR2 (7.03%) but better against PI3Kα (38.77%). Furthermore,compounds 6f,6i and 6j exhibit moderate inhibitory activities against VEGFR2 kinase within the range of 26%-36% and good PI3Kα inhibitory activity in the range of 38%- 58%. This result is consistent with cell cytotoxicity activity. Our findings suggest that compounds 6i and 6j exhibit good PI3Kα inhibitory activity and moderate VEGFR-2 inhibitory activities, while compound 6f showed moderate PI3Kα inhibitory activity and slightly weaker VEGFR-2 inhibitory activity.
|
|
Table 3 Inhibitory activity of compounds selected with two kinases (inhibitory rate,%) at 20 μmol/L. |
{kind=link}
{kind=link}
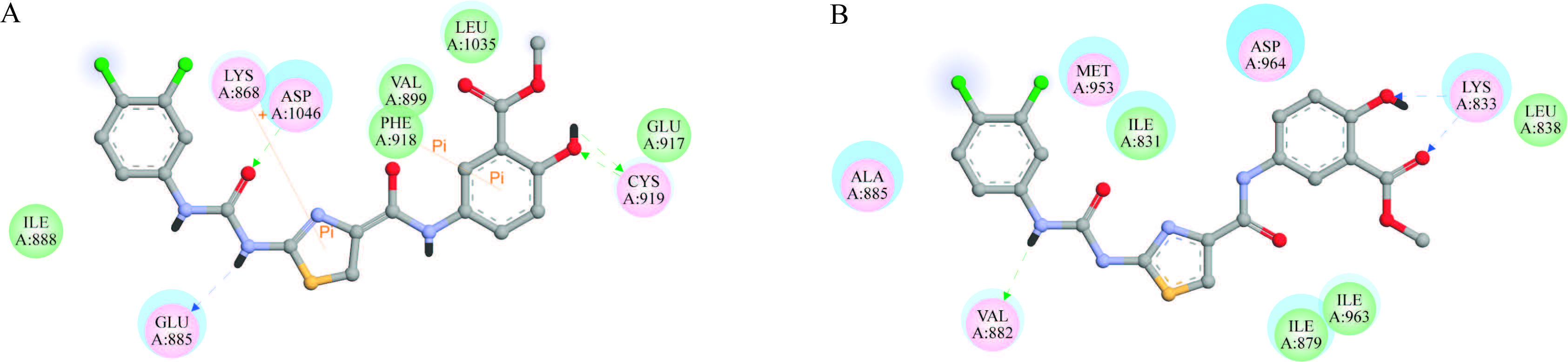
In order to better understand the interaction between compounds and VEGFR and PI3K kinases,compound 6i was selected as a representative example for this series of compounds for molecular docking studies using the Discovery Studio 3.1/ CDOCKER protocol [18, 19]. The VEGFR-2 docking study revealed that the 6i formed three strong hydrogen bonds,one π-π interaction,and one cation-π interaction between the binding site and the ligand (Fig. 1A). Two hydrogen bonds were formed between phenolic hydroxyl and CYS919,and the other one was formed between the urea hydrogen atom and GLU885. The π-π interaction and cation-π interaction were formed with PHE918 and LYS868,respectively.

|
Download:
|
| Fig. 1.2D-presentation for the binding interactions of compound 6i with VEGFR and PI3K kinase domain. (A) Compound 6i with VEGFR; (B) compound 6i with PI3K. | |
{kind=link}
Compound 6i was also docked onto the PI3K-binding domain. Fig. 1B demonstrates that the ligand 6i formed three hydrogen bonds with the protein. The phenolic hydroxyl oxygen atom and carbonyl oxygen atom as hydrogen bond acceptor formed two hydrogen bonds with LYS833. Also the urea group as hydrogen bond donors formed one hydrogen bond with VAL882. The docking analysis indicated that compound 6i fit into the binding site of VEGFR and PI3K kinases suggesting that this compound may be a potent VEGFR and PI3K inhibitor.
The docking analysis indicated that compound 6i fit into the binding site of VEGFR and PI3K kinases suggesting that this compound may be a potent VEGFR and PI3K inhibitor.
4. ConclusionIn summary,a series of novel compounds possessing a 2- aminothiazole scaffold were prepared. Most of the modified compounds showed tantamount or better cytotoxicity against either HepG2 or MDA-MB-231 cell lines. Compounds 6h-6l showed higher and better potency against the two cancer cell lines than Sorafenib. SARs studies indicated that a benzene ring and thiazole ring linked by urea,and as well as removal of the methane group,are crucial for the antitumor activity. The molecular docking studies and the results of kinase inhibition assay in vitro suggest that compound 6i may be a potent VEGFR-2 and PI3Kα dual inhibitor. The 2-aminothiazole scaffold may be considered a promising structure for future designs of VEGFR-2 and PI3Kα dual inhibitors.
AcknowledgmentsThe authors would like to thank the financial supports from the NSFC (No. 21272134) and ShenzhenMunicipal government SZSITIC (Nos. JCYJ20130402145002384,ZDSY20120619141412872).
Appendix A. Supplementary dataSupplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2015.09.008.
| [1] | M. Abdelrahim, S. Konduri, R. Basha, et al., Angiogenesis:an update and potential drug approaches(review), Int. J. Oncol. 36(2010) 5-18. |
| [2] | P. Bhargava, M.O. Robinson, Development of second-generation VEGFR tyrosine kinase inhibitors:current status, Curr. Oncol. Rep. 13(2011) 103-111. |
| [3] | P. Carmeliet, R.K. Jain, Angiogenesis in cancer and other diseases, Nature 407(2000) 249-257. |
| [4] | K.M. Cook, W.D. Figg, Angiogenesis inhibitors:current strategies and future prospects, CA-Cancer J. Clin. 60(2010) 222-243. |
| [5] | P.S. Sharma, R. Sharma, T. Tyagi, VEGF/VEGFR pathway inhibitors as anti-angiogenic agents:present and future, Curr. Cancer Drug Targets 11(2011) 624-653. |
| [6] | J.A. Engelman, J. Luo, L.C. Cantley, The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism, Nat. Rev. Genet. 7(2006) 606-619. |
| [7] | M.T. Burger, S. Pecchi, A. Wagman, et al., Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 Kinase inhibitor for treating cancer, ACS Med. Chem. Lett. 2(2011) 774-779. |
| [8] | Y.W. Zhao, L. Jin, Z.M. Li, et al., Enhanced antitumor efficacy by blocking activation of the phosphatidylinositol 3-kinase/Akt pathway during anti-angiogenesis therapy, Cancer Sci. 102(2011) 1469-1475. |
| [9] | G.D. Thakker, D.P. Hajjar, W.A. Muller, et al., The role of phosphatidylinositol 3-kinase in vascular endothelial growth factor signaling, J. Biol. Chem. 274(1999) 10002-10007. |
| [10] | G.R. Gao, J.L. Liu, D.S. Mei, et al., Design, synthesis and biological evaluation of acylhydrazone derivatives as PI3K inhibitors, Chin. Chem. Lett. 26(2015) 118-120. |
| [11] | S. Brader, S.A. Eccles, Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis, Tumori 90(2004) 2-8. |
| [12] | X.H. Ma, R. Wang, C.Y. Tan, et al., Virtual screening of selective multitarget kinase inhibitors by combinatorial support vector machines, Mol. Pharm. 7(2010) 1545-1560. |
| [13] | L.Y. Han, X.H. Ma, H.H. Lin, et al., A support vector machines approach for virtual screening of active compounds of single and multiple mechanisms from large libraries at an improved hit-rate and enrichment factor, J. Mol. Graph. Model. 26(2008) 1276-1286. |
| [14] | D.M. Zhao, W.Y. Li, Y.F. Shi, et al., Pharmacophore-based design, synthesis, and biological evaluation of novel 3-((3,4-dichlorophenyl)(4-substituted benzyl)amino)propanamides as cholesteryl ester transfer protein(CETP) inhibitors, Chin. Chem. Lett. 25(2010) 299-304. |
| [15] | F. Palagiano, L. Arenare, E. Luraschi, et al., ChemInform abstract:research on heterocyclic compounds, Part 34. Synthesis and SAR study of some imidazo(2,1-b)thiazole carboxylic and acetic acids with antiinflammatory and analgesic activities, ChemInform 27(1996). |
| [16] | A.J. King, A.S. Judd, A.J. Souers, Inhibitors of Diacylglycerol Acyltransferase:a review of 2008 patents, Expert. Opin. Ther. Pat. 20(2010) 19-29. |
| [17] | D.M. Swanson, C.R. Shah, B. Lord, et al., Heterocyclic replacement of the central phenyl core of diamine-based histamine H3 receptor antagonists, Eur. J. Med. Chem. 44(2009) 4413-4425. |
| [18] | M.H. Potashman, J. Bready, A. Coxon, et al., Design, synthesis, and evaluation of orally active benzimidazoles and benzoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors, J. Med. Chem. 50(2007) 4351-4373. |
| [19] | S.D. Knight, N.D. Adams, J.L. Burgess, et al., Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin, ACS Med. Chem. Lett. 1(2010) 39-43. |