 2015, Vol.26
2015, Vol.26 



Since carbocyclic nucleosides have been proven to have a wide range of biological activities,several approaches to the synthesis of these compounds and their analogues have been established over the past decades [1, 2, 3, 4]. 4-Aminocyclopent-2-en-1-ol has great potential as a chiral building block of 50-norcarbocyclic nucleosides [5, 6] and 1-amino-4-hydroxymethyl-2-cyclopentenes have been reported to be versatile precursors for novel carbocyclic nucleosides [7]. Ticagrelor,displaying antiplatelet activity,has also been reported to be derived from 4-aminocyclopent-2-en-1-ol by peripheral elaboration [8, 9].
Compared with conventional chemical methods,biological synthesis of optical active carbocyclic nucleosides is more stereospecific, environmentally friendly and efficient. Although the biological synthesis of typical 5'-(hydroxymethyl) cyclopentane derivatives [10, 11] and 2-cyclopentene-1,4-diol derivatives [12, 13, 14] have been explored and discussed in many published reports, insufficient research has been targeted at 4-aminocyclopent-2-en- 1-ol. Limited biological methods are available to give access to structurally diverse carbocyclic nucleosides,such as polyoxins and nikkomycins [15]. Therefore,it is necessary to devise efficient and inexpensive biocatalytic processes for 4-aminocyclopent-2-en-1-ol precursors. Miller et al. [16] has reported a biocatalytic route by using Candida antarctica lipase B and Pseudomonas species lipase to catalyze the acetylation of cis-N-(benzylcarbamoyl)-4-aminocyclopent- 2-en-1-ol to give the corresponding acetate moiety with (1S,4R) configurationin90%and92%ee,respectively. Inspiredbythis model and aiming toimprove opticalpurity of the target product,we started to explore how CALB acted on other analogues.
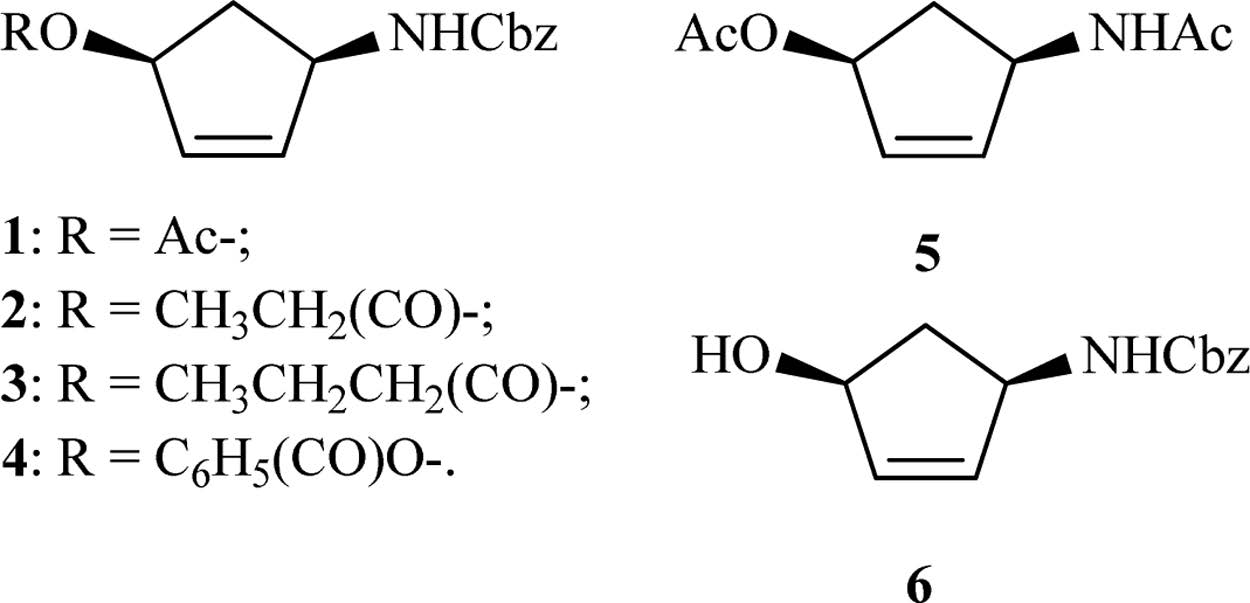
C. antarctica lipase B (CALB),also called Novozyme 435 (commercial name of corresponding immobilized enzyme),is a widely used enzyme with strong hydrolysis and acylation activities towards a variety of esters,amides and thiols,and exhibits a high degree of selectivity [17]. Herein,a number of substrates were studied (Fig. 1),and revealed the chiral hydrolysis activities of CALB towards several N-(benzylcarbamoyl)-4-aminocyclopent- 2-en-1-ol precursors.

|
Download:
|
| Fig. 1.Structures of 4-aminocyclopent-2-en-1-ol analogues. | |
The CALB (GenBank accession No. Z30645.1) studied in this research was produced by genetic engineering with Pichia Pastoris as a host organism. For the extracellular expression of CALB,the gene was amplified by using the forward primer (5'-CCGGAATTCCTACCTTCCGGTTCGGACCCTGCC- 3') and the reverse primer (5'- AAGGAAAAAAGCGGCCGCTCAGG- GGGTGACGATGCCG-3') from pMD18T-CALB construct,with EcoR I and Not I restriction sites introduced,respectively. The PCR product was digested with EcoR I and Not I and then ligated to the vector pPIC9K,which was digested with the same restriction enzymes. The correct linear pPIC9KCALB construct,obtained after digested with Sal I restriction enzyme,was inserted into the host genome through electrotransformationmeans of 1500 V and 8 ms. Integration of CALB into the genome of P. Pastoris in AOX I or His 4 site was verified by PCR analysis using vector- and genome-specific primers,and positive cloneswere further selected by G418 resistance.With the regular addition of methanol to activate the alcohol oxidase in P. Pastoris under the condition of 28 °C and 220 rpm for 168h in BMMY medium.
Several substrates (Fig. 1) were prepared for enzymatic resolution. Compound 1 was prepared by four steps from hydroxylamine hydrochloride and cyclopentadiene with the catalysis of Mo(CO)6 according to the literature procedures [16- 18]. Compounds 2,3 and 4 were prepared based on literature procedure by replacing Ac2O with propionyl chloride,butyryl chloride,and benzoyl chloride,respectively,at the last step and were subjected to 1HNMR analysis to confirm their structures [19]. Compound 5 (Fig. 1) was prepared as a white solid by a slightly modified method [16, 17, 18, 19, 20].
The hydrolysis reaction with CALB was first performed in a closed centrifuge tube,which was initiated with 1 g/L of compound and 0.4 mg/mL CALB suspended in 1 mL phosphate buffer pH 7.0. After incubation at 60 °C for 4 h,the reaction solution was further extracted with 1 mL isopropyl ether. Finally,the isopropyl ether extract (10 μL) was applied to HPLC assay [21].
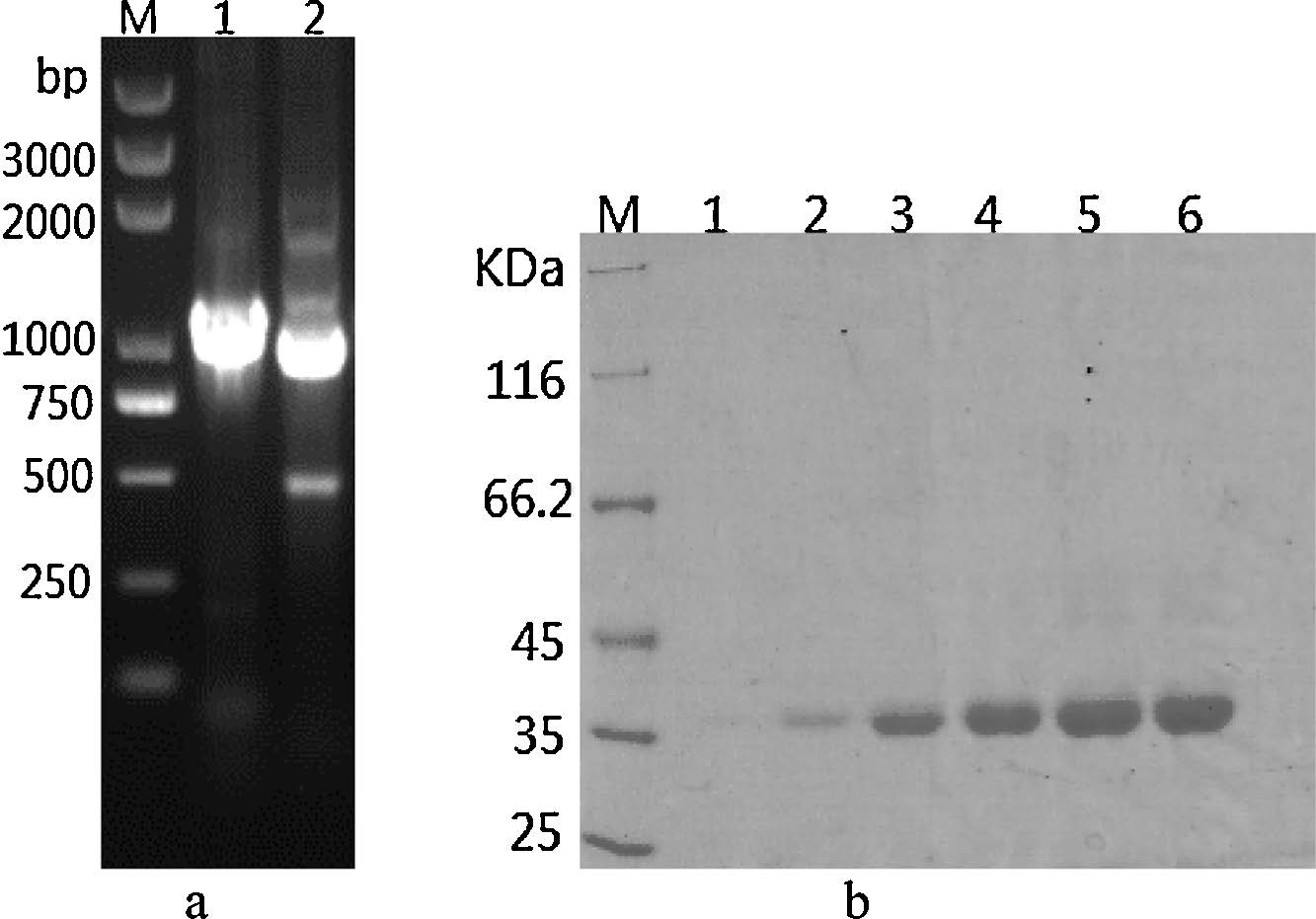
3. Results and discussionThe PCR analysis result using vector- and genome-specific primers was as follows (Fig. 2a). As expected,with the regular addition of methanol to activate the alcohol oxidase in P. Pastoris, the target protein was successfully expressed and accumulated and finally precipitated at 50% ammonium sulfate saturation (Fig. 2b).

|
Download:
|
| Fig. 2.(a) Agarose gel electrophoresis of PCR analysis using vector- and genomespecific primers. M, DNA marker 8000; lane 1, PCR product by using vector-specific primers 3'AOX I and a-factor; lane 2, PCR product by using genome-specific primers. (b) Coomassie-stained gel after SDS-PAGE analysis of supernatant proteins expressed from Pichia Pastoris after purification by 50% ammonium sulfate precipitation. M, protein marker; lanes 1–6, supernatant samples collected at day 2 to day 7, respectively. | |
Initially,compound 3 was chosen as model substrate to optimize reaction conditions. As temperature can significantly influence the solubility of the substrate and activity of the enzyme, we first investigated the effect of temperature on the conversion. Reactions were initiated with 1 g/L of compound 3 and 0.4 mg/mL CALB suspended in 1 mL phosphate buffer pH 7.0,then shaken at different temperatures ranging from 20 °C to 70°C. The results showed that the rate of reaction was significantly improved at 50-60 °C,and then decreased as temperature continued to rise. The optimal reaction pH was subsequently studied at 60 °C. The reaction was carried out from pH 3.0 to pH 9.0. It was found that the reaction was faster under weak acidic conditions,and the optimal pH value was 5.0. Optimal enzyme concentrations and reaction times were further investigated to reduce the spontaneous hydrolysis of substrates. The results showed that the optimal enzyme concentration was about 0.4 mg/mL. However,the optimal reaction time was different for different substrates,and the results were listed in Table 1.
| Table 1 Biocatalysis of different substrates by CALB.a |

{kind=link}
{kind=link}
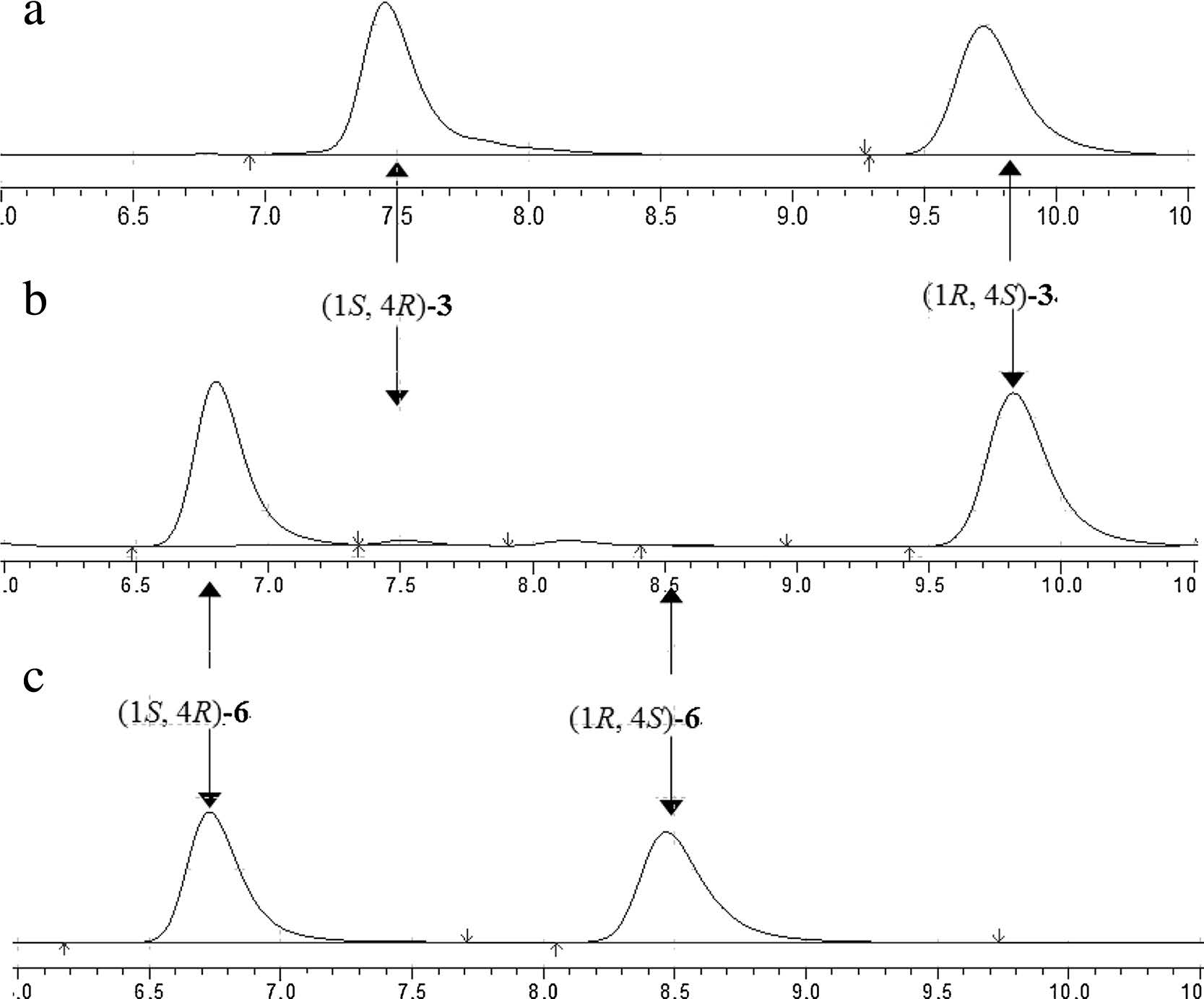
The reaction extract of compound 3 was assayed by HPLC. As shown in Fig. 3,CALB performs an excellent enantioselectivity with racemic 3,and gives the target product (1S,4R)-6 in a yield of 45.7% and an enantiomeric excess of 99.5%.

|
Download:
|
| Fig. 3.(a) Chiral HPLC analysis of racemic 3. (b) Chiral HPLC analysis of the reaction extract, and product 6 with the target configuration (1S,4R) was synthesized. (c) Chiral HPLC analysis of racemic 6. | |
{kind=link}
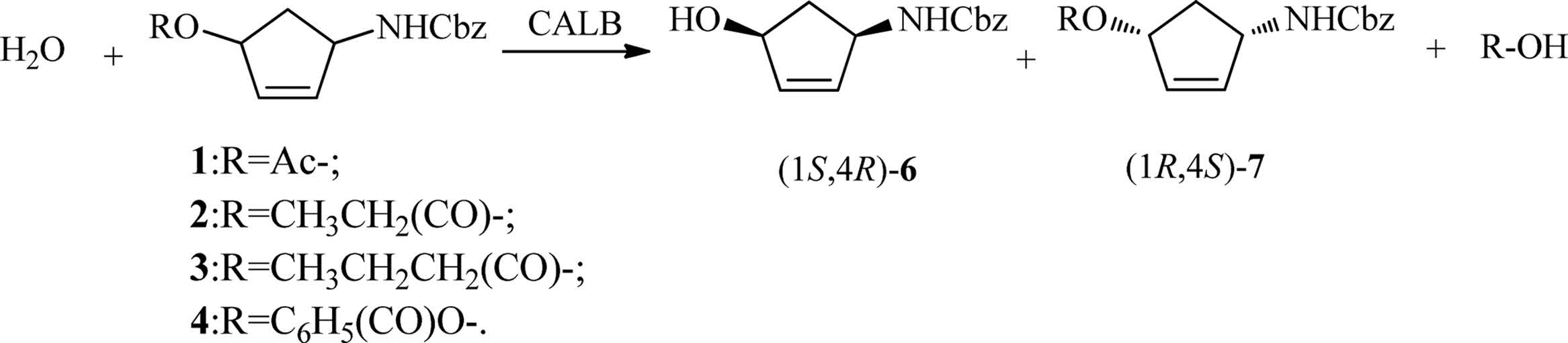
The research was finally conducted under optimal conditions as mentioned above. The results revealed that CALB showed excellent enantioseletivity with compounds 1,2 and 3. Conversion of compounds 1,2 and 3 gave the target product 6 in 99.3%, 98.8% and 99.5% ee,respectively. However,the spontaneous hydrolysis of these substratesmade it difficult to obtain product 6 in high yield. Those results are summarized in Table 1. Subsequently, as shown in Scheme 1,when the reaction time was lengthened to 8 h,we obtained (1S,4R)-6 in >94.0% ee with the yield up to 42.0%. At the same time,the ester 7was producedwith approximately 99.0% ee and the yield up to 43.0% which may be used for other applications.

|
Download:
|
| Scheme. 1.Enzymatic hydrolysis to obtain (1S,4R)-N-(benzylcarbamoyl)-4-aminocyclopent-2-en-1-ol by CALB. | |
{kind=link}
In summary,an efficient biocatalytic process to obtain optically active (1S,4R)-N-(benzylcarbamoyl)-4-aminocyclopent-2-en-1-ol was developed by employing CALB as the catalyst and using compound 1,2,3,4 or 5 as substrate. Compared with the results reported by Miller [16],we obtained the target product (1S,4R)-6 with substrates 1,2,3 with above 98.0% ee and the yield up to 45.0%. In addition,we found CALB may not be an appropriate enzyme to catalyze compounds 4 and 5,which implied that the length of the side chain may have an effect on the enantioselectivity [17]. Taken together,our work has provided an approach to improve the purity of the target product,(1S,4R)-N-(benzylcarbamoyl)- 4-aminocyclopent-2-en-1-ol,which can be used as a chiral intermediate of ticagrelor and other important chiral drugs.
AcknowledgmentThis work was financially supported by the Fundamental Research Funds for the Central Universities (No. YS1407).
| [1] | F. Burlina, A. Favre, J.L. Fourrey, M. Thomas, An expeditious route to carbocyclic nucleosides: (61)-aristeromycin and (61)-carbodine, Bioorg. Med. Chem. Lett. 7 (1997) 247-250. |
| [2] | K.H. Park, H. Rapoport, Enantioselective synthesis of (1R,4S)-1-amino-4-(hydroxymethyl)- 2-cyclopentene, a precursor for carbocyclic nucleoside synthesis, J. Org. Chem. 59 (1994) 394-399. |
| [3] | B.M. Trost, D. Stenkamp, S.R. Pulley, An enantioselective synthesis of cis-4-tertbutoxycarbamoyl- 1-methoxycarbonyl-2-cyclopentene—a useful, general building block, Chem. A: Eur. J. 1 (1995) 568-572. |
| [4] | T. Ando, K. Kojima, P. Chahota, et al., Synthesis of 4'-modified noraristeromycins to clarify the effect of the 4'-hydroxyl groups for inhibitory activity against S-adenosyl-L-homocysteine hydrolase, Bioorg. Med. Chem. Lett. 18 (2008) 2615-2618. |
| [5] | N.G. Ramesh, A.J. Klunder, B. Zwanenburg, Enantioselective synthesis of 4-acetylaminocyclopent- 2-en-1-ols from tricyclo[5.2.1.0(2, 6)]decenyl enaminones. Precursors for 5'-norcarbocyclic nucleosides and related antiviral compounds, J. Org. Chem. 64 (1999) 3635-3641. |
| [6] | P.F. Vogt, J.G. Hansel, M.J. Miller, Asymmetric synthesis of an important precursor to 5'-nor carbocyclic nucleosides, Tetrahedron Lett. 38 (1997) 2803-2804. |
| [7] | D. Zhang, A. Ghosh, C. Suling, et al., Efficient functionalization of acylnitroso cycloadducts: application to the syntheses of carbocyclic nucleoside precursors, Tetrahedron Lett. 37 (1996) 3799-3802. |
| [8] | J.J.J. Van Giezen, L. Nilsson, P. Berntsson, et al., Ticagrelor binds to human P2Y (12) independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation, J. Thromb. Haemostasis. 7 (2009) 1556-1565. |
| [9] | B. Springthorpe, A. Bailey, P. Barton, From ATP to AZD6140: the discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis, Bioorg. Med. Chem. Lett. 17 (2007) 6013-6018. |
| [10] | M. Arita, K. Adachi, Y. Ito, et al., Enantioselective synthesis of the carbocyclic nucleosides (-)-aristeromycin and (-)-neplanocin A by a chemicoenzymatic approach, J. Am. Chem. Soc. 105 (1983) 4049-4055. |
| [11] | M. Ikbal, C. Cerceau, F. Le Goffic, et al., Synthè se des deux énantiomè res de l'analogue carbocyclique du nicotinamide ribose et évaluation de leurs proprié té s biologiques, Eur. J. Med. Chem. 24 (1989) 415-420. |
| [12] | S. Saul, S. Corr, J. Micklefield, Biotransformations in low-boiling hydrofluorocarbon solvents, Angew. Chem. Int. Ed. 43 (2004) 5519-5523. |
| [13] | J. Dauvergne, A.M. Happe, V. Jadhav, et al., Synthesis of 4-azacyclopent-2-enones and 5,5-dialkyl-4-azacyclopent-2-enones, Tetrahedron 60 (2004) 2559-2567. |
| [14] | C.R. Johnson, S.J. Bis, Enzymatic asymmetrization of meso-2-cycloalken-1 4-diols and their diacetates in organic and aqueous media, Tetrahedron Lett. 33 (1992) 7287-7290. |
| [15] | D. Zhang, M.J. Miller, Total synthesis of (±) carbocyclic polyoxin C and its α-epimer, J. Org. Chem. 63 (1998) 755-759. |
| [16] | M.J. Mulvihill, J.L. Gage, M.J. Miller, Enzymatic resolution of aminocyclopentenols as precursors to D-and L-carbocyclic nucleosides, J. Org. Chem. 63 (1998) 3357-3363. |
| [17] | E.M. Anderson, K.M. Larsson, O. Kirk, One biocatalyst - many applications: the use of Candida antarctica B-lipase in organic synthesis, Biocatal. Biotransform. 16 (1998) 181-204. |
| [18] | L. Bollans, J. Bacsa, J.A. Iggo, et al., The acyl nitroso Diels-Alder (ANDA) reaction of sorbate derivatives: an X-ray and 15N NMR study with an application to aminoacid synthesis, Org. Biomol. Chem. 7 (2009) 4531-4538. |
| [19] | Compound 2: 1H NMR (400 MHz, CDCl3): δ 7.47-7.09 (m, 5H), 5.97 (m, 2H), 5.55 (s, 1H), 5.11 (s, 2H), 4.85 (m, 1H), 4.76 (m, 1H), 2.84 (m, 1H), 2.30 (t, 2H), 1.56 (m, 1H), 1.17 (t, 3H). Compound 3: 1H NMR (400 MHz, CDCl3): d 7.61-7.24 (m, 5H), 5.98 (m, 2H), 5.55 (s, 1H), 5.11 (s, 2H), 4.83 (m, 1H), 4.69 (m, 1H), 2.97-2.65 (m, 1H), 2.29 (t, 2H), 1.70-1.48 (m, 3H), 0.98 (t, 3H). Compound 4: 1H NMR (400 MHz, CDCl3): δ 7.94-7.13 (m, 10H), 6.01 (m, 2H), 5.72 (s, 1H), 5.11 (s, 2H), 4.85(m, 1H), 4.75 (m, 1H), 2.97-2.85 (m, 1H), 1.65 (m, 1H). |
| [20] | L. Werner, J.R. Hudlicky, M. Wernerova, et al., Synthesis of 1,2-and 1,4-amino alcohols from 1,3-dienes via oxazines. Rearrangements of 1,4-amino alcohol derivatives to oxazolines, Tetrahedron 66 (2010) 3761-3769. |
| [21] | Analytical HPLC methods: the enantiomeric excess analysis was performed using a HPLC system (Shimadzu, Japan) equipped with a CHIRALPAK IF column (0.46 mm × 250 mm × 5 mm), elution with ethanol/hexane 15:85 v/v for compound 1, ethanol/hexane 25:75 v/v for compounds 2 and 3, ethanol/hexane 30:70 for compound 4. |