 2015, Vol.26
2015, Vol.26 



b State Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
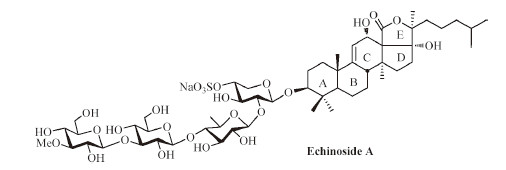
Sea cucumber saponins are common secondary metabolites produced by holothurian species,which play an important role in the chemical defense of the slow-moving animals while show a wide spectrum of pharmacological activities. Echinoside A,a lanostane-type triterpene oligosaccharide,was isolated from the sea cucumber Actinopyga echinites (JAEGER) by Kitagawa et al. [1] (Fig. 1). This compound demonstrated potent activities in vitro and in vivo against a broad-spectrum of antitumor cells,especially it could directly kill P-gp-mediated multidrug-resistant tumor cells [2].

|
Download:
|
| Fig. 1.The structure of Echinoside A. | |
{kind=link}
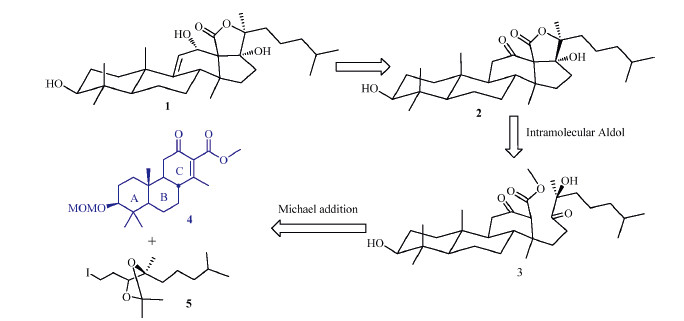
Echinoside A consists of a linear tetrasaccharide attached to a pentacyclic triterpene aglycon. The construction of the pentacyclic triterpene aglycon constitutes the most challenging part of the synthesis; in fact,the synthesis of Echinoside A has never been reported so far [3]. As shown in Scheme 1,we envisaged to construct the pentacyclic aglycon (1) by introduction of the double bond at the a position of the ketone in skeleton 2,followed by reduction of the ketone. Compound 2 could be constructed via an intramolecular Aldol reaction from compound 3,which would be synthesized through a Michael addition between the terminal alkyl iodide 5 and the ABC skeleton 4. Herein,we report the synthesis of the key ABC skeleton intermediate (4).
|
|
Download:
|
| Scheme 1.Retrosynthetic analysis of the aglycon 1. | |
Commercial reagents were used without further purification unless specialized. Solvents were dried and redistilled prior to use in the usual way. Thin layer chromatography (TLC) was performed on precoated plates of Silica Gel HF254 (0.2 mm,Yantai,China). Flash column chromatography was performed on Silica Gel H (10-40 m,Yantai,China). Optical rotations were determined with a Perkin-Elmer Model 241 MC polarimeter. 1H NMR and 13C NMR spectra were recorded on a Bruker AM 400 spectrometer with Me4Si as the internal standard. Chemical shifts were recorded in δ values and J values were given in Hz. Mass spectra were obtained on a HP5989A or a VG Quatro mass spectrometer.
Wieland-Miescher ketone 9: A suspension of ketone 6 (25.0 g,198.18 mmol) in water (60 mL) was treated with HOAc (0.6 mL),hydroquinone (225 mg,2.043 mmol),and fresh distilled methyl vinyl ketone (32 mL,387.10 mmol). Refluxed at 80 ℃ overnight,returned to the room temperature,NaCl (20.0 g) was added,then the mixture was diluted with EtOAc. The solution was washed with brine,dried with Na2SO4,and then concentrated to afford the crude product 8 (30.15 g,78%).
In a standard glass vial with stirrer bar was added triketone 8 (6.14 g,31.29 mmol) followed by the catalyst 10 (335 mg,0.625 mmol) and benzoic acid (20 mg,0.164 mmol). The resulting mixture darkened and was stirred for 7 days. The mixture was absorbed onto silica gel and purified by column chromatography (petroleum ether/EtOAc 3:1-1:1) to give the Wieland-Miescher ketone 9 (5.19 g,93%,90% ee) as a clear oil. Recrystallization from Et2O gives a white crystalline solid (99.5% ee) [4]. 1H NMR mmol) were added to a solution of 9 (2.10 g,11.783 mmol) in ethylene glycol (40 mL). After being stirred at room temperature for 30 min,the reaction mixture was poured slowly into a 2:1 mixture of ice-water/sat. aqueous NaHCO3. The mixture was extracted with EtOAc four times,and the combined organic layers were washed with brine,dried over Na2SO4. Concentration and purification by silica gel column chromatography (petroleum ether/EtOAc = 6:1) afforded 11 (2.28 g,87%) as a yellow oil [5]. 1H NMR (400 MHz,chloroform-d): δ 5.82 (d,1H,J = 1.9 Hz),4.07-3.85 (m,4H),2.41 (ddd,2H,J = 10.9,4.8,3.0 Hz),2.35-2.24 (m,2H),1.96-1.85 (m,1H),1.85-1.59 (m,5H),1.36 (s,3H).
Compound 11: 4 Å molecular sieves (2.5 g) and p-TsOH.H2O (2.20 g,11.566 mmol) were added to a solution of 9 (2.10 g,11.783 mmol) in ethylene glycol (40 mL). After being stirred at room temperature for 30 min,the reaction mixture was poured slowly into a 2:1 mixture of ice-water/sat. aqueous NaHCO3. The mixture was extracted with EtOAc four times,and the combined organic layers were washed with brine,dried over Na2SO4. Concentration and purification by silica gel column chromatography (petroleum ether/EtOAc = 6:1) afforded 11 (2.28 g,87%) as a yellow oil [5]. 1H NMR (400 MHz,chloroform-d): δ 5.82 (d,1H,J = 1.9 Hz),4.07-3.85 (m,4H),2.41 (ddd,2H,J = 10.9,4.8,3.0 Hz),2.35-2.24 (m,2H),1.96-1.85 (m,1H),1.85-1.59 (m,5H),1.36 (s,3H).
Compound 12: A solution of 11 (2.28 g,10.257 mmol) in n-propanol (20 mL),was treated with PhSH (1.6 mL,15.521 mmol),37% HCHO (1.26 mL,16.555 mmol),Et3N (1.4 mL,15.521 mmol),and HCOOK (1.044 g,12.416 mmol) at 100℃ for 24 h. Then the mixture was diluted with CH2Cl2,and the solution was washed with saturated aqueous NaHCO3 and brine,dried with Na2SO4. Concentration and purification by silica gel column chromatography (petroleum ether/EtOAc 8:1) afforded 12 (2.86 g,81%) as yellow crystals [6]. 1H NMR (400 MHz,chloroform-d): δ 7.43-7.36 (m,2H),7.30-7.17 (m,3H),4.02-3.89 (m,5H),3.76 (d,1H,J = 11.5 Hz),2.66 (ddt,1H,J = 15.5,4.2,2.0 Hz),2.49 (dt,1H,J = 16.0,4.4 Hz),2.37 (ddd,1H,J = 16.0,13.9,4.9 Hz),2.24 (td,1H,J = 13.5,4.9 Hz),2.03 (td,1H,J = 14.6,5.6 Hz),1.85 (td,1H,J = 13.5,4.5 Hz),1.75 (ddt,1H,J = 10.2,4.5,2.5 Hz),1.70-1.62 (m,2H),1.57-1.50 (m,1H),1.32 (s,3H).
Compound 13: A solution of 12 (6.30 g,18.79 mmol) in THF/tBuOH (100 mL/3.5 mL),was added to a stirred solution of 3 equiv. of lithium in liquid ammonia (500 mL) over a 20-30 min period. The solution was allowed to stir at -72℃ for another 45 min,whereupon 100 mL of THF was added followed by rapid addition of CH3I (17.1 mL,274.335 mmol). The mixture was allowed to stir for 30 min after which the ammonia was allowed to evaporate. The mixture was concentrated and the residue was diluted with CH2Cl2 and washed with water,brine,dried over Na2SO4,and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 25:1) to afford 13 (2.86 g,81%) as colorless oil [7]. 1H NMR (500 MHz,chloroformd): δ3.98-3.83 (m,4H),2.62 (ddd,1H,J = 15.1,13.6,6.3 Hz),2.33 (ddd,1H,J = 15.2,5.3,3.5 Hz),1.99-1.90 (m,1H),1.89-1.83 (m,1H),1.77-1.64 (m,3H),1.55-1.43 (m,4H),1.24 (s,3H),1.08 (s,3H),1.04 (s,3H).
Compound 14: A solution of 13 (1.23 g,4.86 mmol) in EtOH (15 mL) was treated with NaBH4 (184 mg,4.86 mmol) at-40℃ for 2 h. Diluted with Et2O and washed with water and brine,the organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 10:1) to afford 14 (1.10 g,89%) as colorless crystal [7]. 1H NMR (400 MHz,chloroform-d): δ 3.98-3.88 (m,3H),3.83 (dd,1H,J = 7.5,3.6 Hz),3.30-3.20 (m,1H),1.74-1.24 (m,12H),1.05 (s,3H),0.99 (s,3H),0.80 (s,3H).
Compound 15: A solution of 14 (500 mg,1.97 mmol) in EtOH/ H2O (18 mL/2 mL),was treated with PPTS (64 mg,0.26 mmol) and refluxed for 2 h. The solvent was concentrated and diluted with CH2Cl2. The mixture was washed with water and brine,dried over Na2SO4,and concentrated,affording the crude product.
The crude product was dissolved in CH2Cl2 (10 mL),and was then treated with DIPEA (1.72 mL,9.83 mmol),MOMCl (0.74 mL,9.83 mmol) at room temperature for 2 h. Quenched with saturated NH4Cl,diluted with CH2Cl2,and washed with water and brine,the resulting organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 15:1) to afford 15 (490 mg,98%) as colorless oil. 1H NMR (400 MHz,chloroform-d): δ 4.74 (d,1H,J = 6.8 Hz),4.60 (d,1H,J = 6.8 Hz),3.39 (s,3H),3.10-3.02 (m,1H),2.57 (td,1H,J = 13.9,7.0 Hz),2.20 (dddd,1H,J = 14.1,5.0,2.2,1.3 Hz),2.13-2.03 (m,1H),1.91-1.83 (m,1H),1.76 (dt,1H,J = 6.7,2.6 Hz),1.74-1.68 (m,1H),1.68-1.53 (m,4H),1.16 (s,3H),1.13 (d,1H,J = 3.3 Hz),1.00 (s,3H),0.92 (s,3H). ESI-MS (m/z): 277.1 [M+Na]+. HRMS (m/z): [M+H]+ calcd. for C15H27O3 255.1955,found 255.1952.
Compound 16: A solution of 15 (1.5 g,5.897 mmol) in dry toluene/THF (15 mL/5 mL),was treated with 60% NaH (472 mg,11.8 mmol) at 0 ℃ for 30 min. HCOOEt (5 mL) was added and stirring continued at room temperature for 4 h. Quenched with saturated NH4Cl,diluted with EtOAc and washed with water and brine,the resulting organic layer was dried over Na2SO4 and concentrated to afford a crude product.
The crude product was dissolved in CH2Cl2 (10 mL) and then treated with MVK (2.0 mL,23.56 mmol) and Et3N (2.5 mL,17.69 mmol). After stirring at room temperature for 4 h,the mixture was concentrated to afford a crude product.
The crude product was dissolved in HOBut (25 mL) and then treated with KOBut (990 mg,8.85 mmol). After stirring at room temperature overnight,the mixture was quenched with water and concentrated. The residue was diluted with EtOAc and washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 6:1) to afford 16 (1.52 g,84%) as colorless oil. 1H NMR (400 MHz,chloroform-d): δ 5.78 (d,1H,J = 1.5 Hz),4.73 (d,1H,J = 6.8 Hz),4.59 (d,1H,J = 6.8 Hz),3.36 (s,3H),3.06 (dd,1H,J = 11.5,4.2 Hz),2.52 (dddt,1H,J = 14.2,7.4,5.3,2.1 Hz),2.35 (dt,1H,J = 16.2,4.9 Hz),2.22 (ddd,1H,J = 16.2,12.7,5.0 Hz),2.10-1.97 (m,2H),1.92-1.84 (m,1H),1.76-1.43 (m,6H),1.25-1.15 (m,1H),1.10 (s,3H),1.04 (dd,1H,J = 12.2,2.8 Hz),0.96 (s,3H),0.86 (s,3H). 13C NMR (101 MHz,chloroform-d): δ 201.23,175.58,119.84,96.04,84.34,55.56,52.45,40.53,39.30,35.92,35.02,34.63,34.19,29.28,28.11,24.20,21.33,
21.09,16.49. ESI-MS (m/z): 329.4 [M+Na]+; HRMS (m/z): [M+Na]+ calcd. for C19H30O3Na 329.2087,found 329.2091.
Compound 17: A solution of 16 (38 mg,0.12 mmol) in dimethoxyethane (2 mL) was treated with 60% NaH (10 mg,0.25 mmol) and dimethyl carbonate (21 mL,0.25 mmol). The mixture was refluxed under nitrogen for 2 h,and then quenched with saturated NH4Cl and diluted with EtOAc. The mixture was washed with water and brine,dried over Na2SO4,and concentrated. The residue was purified by preparative thin-layer chromatography (petroleum ether/EtOAc 4:1) to afford 17 (5 mg) as colorless oil. 1H NMR (400 MHz,chloroform-d): δ 6.83 (d,1H,J = 8.1 Hz),6.63 (d,1H,J = 2.6 Hz),6.50 (dd,1H,J = 8.2,2.6 Hz),4.72 (d,1H,J = 6.9),4.60-4.56 (d,1H,J = 6.9),3.35 (s,3H),3.09 (dd,1H,J = 11.7,4.4 Hz),2.81 (ddd,1H,J = 16.7,6.7,1.8 Hz),2.76-2.64 (m,1H),2.20-2.10 (m,1H),1.90-1.74 (m,2H),1.74-1.58 (m,2H),1.48-1.34 (m,1H),1.28-1.16 (m,1H),1.12 (s,3H),0.98 (s,3H),0.84 (s,3H). ESI-MS (m/z): 327.4 [M+Na]+.
Compound 18: A solution of 16 (60 mg,0.196 mmol) in THF/ EtOH (3 mL/0.03 mL) was added dropwise to a stirred liquid ammonia (10 mL) at -72 ℃,and then lithium (60 mg) was added to the solution. The solution was allowed to stir for 4 h at -72 ℃. Quenched with saturated NH4Cl after which the ammonia was allowed to evaporate. The mixture was diluted with EtOAc and washed with water and brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 6:1) to afford 18 (1.52 g,84%) as colorless oil. [a]D 25 21.4 (c 0.50,CHCl3); 1H NMR (400 MHz,chloroform-d): δ 4.72 (dd,1H,J = 6.8,1.4 Hz),4.59 (dd,1H,J = 6.8,1.5 Hz),3.37 (s,3H),3.11-3.00 (m,1H),2.29 (dq,3H,J = 11.6,6.6,5.1 Hz),2.03 (t,1H,J = 13.4 Hz),1.99-1.92 (m,1H),1.88 (dt,1H,J = 12.2,3.6 Hz),1.74 (m,1H),1.67-1.46 (m,4H),1.46-1.32 (m,1H),1.32-1.17 (m,1H),1.13-1.04 (m,1H),1.02-0.98 (m,1H),0.98-0.94 (m,1H),0.95 (s,3H),0.88 (s,3H),0.85 (d,1H,J = 2.0 Hz),0.82 (s,3H). 13C NMR (101 MHz,chloroform-d):δ213.12,96.01,84.85,55.77,55.55,54.25,41.13,40.95,38.63,36.81,36.59,35.31,34.10,34.01,28.29,24.09,21.05,
16.53,13.82. ESI-MS (m/z): 331.3 [M+Na]+. HRMS (m/z): [M+Na]+ calcd. for C19H32NaO3:331.2244,found 331.2244.
Compound 19: To a mixture of 60% NaH (28 mg,0.93 mmol) and 30% KH (15 mg,0.11 mmol) in dry THF (0.5 mL) was added 18 (48 mg,0.16 mmol) in dry THF (2 mL) and dimethyl carbonate (33 mL,0.40 mmol) at 0 ℃. The temperature was raised to 70 ℃ and the mixture refluxed for 4 h. After cooling down to 0 ℃,the mixture was acidized with 50% HOAc,and diluted with EtOAc. The mixture was washed with water,aq. NaHCO3,and brine,respectively. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 15:1) to afford 19 (38 mg,67%) as colorless oil. [a]D 25 23.0 (c 0.70,CHCl3); 1H NMR (400 MHz,chloroform-d): δ 12.05 (s,1H),4.73 (t,1H,J = 5.1 Hz),4.60 (dd,1H,J = 6.9,2.6 Hz),3.73 (s,3H),3.38 (s,3H),3.09 (dd,1H,J = 11.7,4.2 Hz),2.39 (dd,1H,J = 15.7,5.2 Hz),2.25-2.05 (m,2H),1.99-1.88 (m,1H),1.80-1.49 (m,6H),1.38 (ddd,2H,J = 16.4,9.5,3.5 Hz),1.04 (td,2H,J = 12.6,5.2 Hz),0.97 (s,3H),0.91-0.88 (m,1H),0.86 (s,3H),0.84 (s,3H). 13C NMR (101 MHz,chloroform-d): δ 172.61,171.77,96.50,95.99,84.97,55.57,54.45,51.37,50.02,38.62,37.18,36.14,34.56,31.87,30.90,28.59,28.35,24.05,
21.00,16.66,13.80. ESI-MS (m/z): 389.4 [M+Na]+. HRMS (m/z): [M+Na]+ calcd. for C21H34NaO5 389.2298,found 389.2300.
Compound 20: A solution of 19 (47 mg,0.13 mmol) in dry CH2Cl2 (2 mL) was treated with pyridine (50 mL) and PhSeCl (50 mg,0.256 mmol) at 0 ℃. The mixture was then stirred at room temperature overnight. Diluted with CH2Cl2 and washed with 1 mol/L HCl and water. The organic layer was added 30% H2O2 (1.5 mL),stirred at room temperature for 10 min until the yellow solution turned to colorless. The organic layer was washed with aq. NaHCO3 and brine,respectively,and was then dried over Na2SO4,and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 6:1) to afford 20 (34 mg,73%) as colorless oil. [a]D 25 16.9 (c 1.0,CHCl3); 1H NMR (500 MHz,chloroform-d): δ 7.40 (d,1H,J = 1.8 Hz),4.73 (d,1H,J = 6.8 Hz),4.59 (d,1H,J = 6.8 Hz),3.78 (s,3H),3.37 (s,3H),3.07 (dd,1H,J = 11.7,4.3 Hz),2.50 (dd,1H,J = 15.8,3.3 Hz),2.44 (tdd,1H,J = 10.6,4.1,1.8 Hz),2.17 (dd,1H,J = 15.7,14.3 Hz),2.09 (dd,1H,J = 13.0,3.5 Hz),1.84-1.72 (m,2H),1.64 (dt,1H,J = 13.0,3.5 Hz),1.60-1.40 (m,3H),1.30-1.21 (m,1H),1.02 (dd,1H,J = 13.3,3.9 Hz),0.96 (s,3H),0.90 (s,3H),0.88 (d,1H,J = 2.5 Hz),0.82 (s,3H). 13C NMR (126 MHz,chloroform-d): δ 195.54,165.07,161.25,131.26,96.04,84.61,55.58,53.97,52.90,52.21,38.79,38.65,37.15,36.21,36.05,32.14,28.06,24.
04,21.42,16.35,14.27. ESI-MS (m/z): 387.3 [M+Na]+; HRMS (m/z): [M+H]+ calcd. for C21H33O5: 365.2323,found 365.2326.
Compound 21: To a solution of 20 (30 mg,0.082 mmol) in dry THF (1 mL) was added a freshly prepared solution of Me2CuLi (0.38 mL,0.09 mmol) in THF. After being stirred at -78 ℃ for 1 h,the mixture was quenched with saturated NH4Cl. The resulting mixture was diluted with EtOAc and washed with water and brine,respectively. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 5:1) to afford 21 (28 mg,89%) as yellow oil. [a]D 25 14.0 (c 1.2,CHCl3); 1H NMR (500 MHz,chloroform-d): δ 12.13 (s,0.13H),4.73 (d,1H,J = 6.7 Hz),4.60 (d,1H,J = 6.8 Hz),3.75 (s,3H),3.38 (s,3H),3.08 (ddd,2H,J = 11.6,4.9,2.5 Hz),2.42 (dd,1H,J = 13.2,3.6 Hz),2.16 (dq,1H,J = 13.2,3.9 Hz),2.13-2.03 (m,1H),1.75 (dt,1H,J = 11.6,4.0 Hz),1.69 (ddd,2H,J = 10.8,4.6,2.3 Hz),1.61-1.47 (m,3H),1.43-1.29 (m,2H),1.27- 1.15 (m,2H),1.11-1.04 (m,1H),0.99 (d,3H,J = 6.4 Hz),0.97 (s,3H),0.95-0.90 (m,1H),0.88 (s,3H),0.83 (s,3H). HRMS (m/z): [M+H]+ calcd. for C22H37O5 381.2636,found 381.2638.
Compound 4: A solution of 21 (822 mg,2.160 mmol) in dry CH2Cl2 (20 mL) was treated with pyridine (0.5 mL) and PhSeCl (827 mg,4.320 mmol) at 0 ℃. After stirring at room temperature overnight,the mixture was diluted with CH2Cl2 and washed with 1 mol/L HCl and water. The organic layer was added 30% H2O2 (2.0 mL),stirred at room temperature for 10 min until the yellow solution turned to colorless. The mixture was washed with aq. NaHCO3 and brine,respectively,and was then dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc 6:1-4:1) to afford 4 (406 mg,49%) as white solid together with 20 (335 mg,41%). 4: [a]D 25 7.56 (c 1.1,CHCl3); 1H NMR (400 MHz,chloroform-d): δ 4.74 (d,1H,J = 6.8 Hz),4.60 (d,1H,J = 6.8 Hz),3.81 (s,3H),3.38 (s,3H),(dd,1H,J = 13.3,3.8 Hz),0.97 (s,3H),0.90 (s,3H),0.83 (s,3H). 13C NMR (101 MHz,chloroform-d): δ 195.99,167.69,162.39,133.01,96.04,84.58,77.24,55.61,53.90,52.25,51.92,39.96,38.57,37.33,36.27,30.09,28.04,
24.13,21.38,19.31,16.35,14.34. ESI-MS (m/z): 401.3 [M+Na]+. HRMS (m/z): [M+H]+ calcd. for C22H35O5: 379.2479,found: 379.2480.
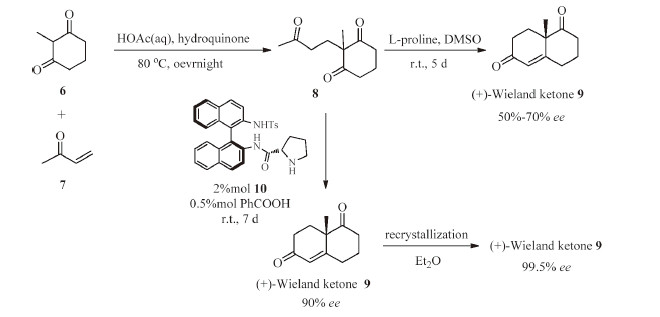
Our synthesis started with the preparation of (+)-Wieland- Miescher ketone (9) through a Robinson annulation reaction (Scheme 2). The Michael addition between 2-methyl-1,3-cyclohexanedione 6 andmethyl vinyl ketone (MVK) 7 provided the triketone 8. Then,the L-proline-promoted aldolization of triketone 8 yielded the Wieland-Miescher ketone 9,albeit in poor enantioselectivity (50-70% ee) [8]. Following the procedure reported by Bonjoch [9],treatment of triketone 8 with N-tosyl-(S)-binam-prolinamide (catalyst 10) through a solvent-free Robinson annulation procedure afforded 9 in high enantioselectivity (90% ee). In addition,the (+)- Wieland-Miescher ketone 9 could be enantiomerically enriched by recrystallization from Et2O (99.5%ee).

|
Download:
|
| Scheme 2.Preparation of (+)-Wieland–Miescher ketone 9. | |
{kind=link}
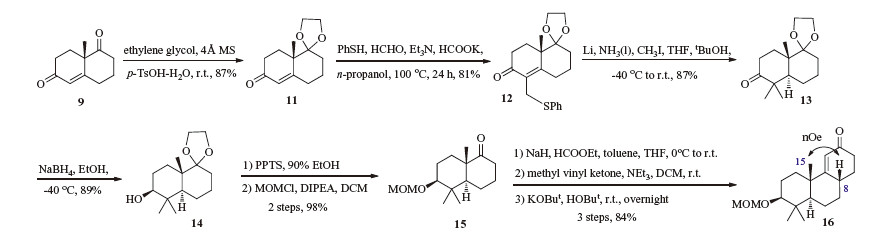
The (+)-Wieland-Miescher ketone 9 was selectively protected as ketal 11,which was subsequently subjected to Kirk-Petrow conditions (Scheme 3) [10]. Indeed,ketal 11 was treated with PhSH,HCHO,Et3N and HCOOK in n-propanol at 100 ℃ for 24 h to furnish the thiol 12 in 81% yield. Reductive alkylation of the enone under Birch conditions provided the 4,4-dimethyl-trans-decalone derivative 13 (87%),which was reduced into the corresponding alcohol 14 by NaBH4 (89%) [11]. Deacetalization followed by protection of the hydroxyl group of 14 with MOMCl afforded the protected ketone 15 in 98% over two steps. Then,the annulation of 15 in order to give the tricyclic enone 16 was achieved under the standard conditions exemplified in Spencer’s work [12]. More precisely,the annulation was accomplished by formylation of the ketone 15,followed by a Robinson annulation employing MVK 7,with concomitant removal of the formyl group under basic conditions. Enone 16 was finally isolated in 84% yield over three steps. The stereochemistry of the C8-H was determined by NOE analysis which exhibits a strong correlation with the C15-H. Other attempts employing 3-penten-2-one and 4-methyl-3-penten-2- one instead of MVK failed to construct the corresponding triketone intermediate via the Robinson annulation.

|
Download:
|
| Scheme 3.Construction of the ABC ring framework. | |
{kind=link}
With the annulation product 16 in hand,we focused our efforts on the installation of the methoxycarbonyl group at the C-13 position (Scheme 4). Treatment of 16 with lithium diisopropylamide (LDA) in -78 ℃,followed by addition of methyl chloroformate led to no product,whereas treatment of 16 with NaH and dimethyl carbonate in dimethoxyethane afforded the aromatization product 17. Consequently,we decided to reduce the C9-C11 double bond at an earlier stage. At first,hydrogenation of 16 was adopted in the presence of catalytic Pd/C,but the desired saturated ketone was isolated in a mixture of the two C9-isomers. Gratifyingly,reduction under Birch conditions yielded the saturated ketone 18 as a single isomer (82%) [13]. Carbomethoxylation was then achieved by heating at 70 ℃ a mixture of 18 and dimethyl carbonate in the presence of NaH and KH in THF. Subsequent phenylselenation and dehydroselenation of 19 provided the desired α,β-unsaturated ketone 20 in 73% yield [14]. Methylation of 20 with Me2CuLi in THF afforded ketone 21 in 89% yield,which was subjected to a similar phenylselenation/ dehydroselenation protocol,thus providing the desired key intermediate 4 in 49% yield (83% yield based on recovered starting materials).

|
Download:
|
| Scheme 4.Synthesis of the key ABC skeleton intermediate 4. | |
{kind=link}
In conclusion,we have achieved the synthesis of the key ABC-fused ring skeleton of the aglycon of Echinoside A (i.e.,4). The synthesis starts from the (+)-Wieland-Miescher ketone and requires 16 steps and in 13% overall yield with Robinson annulation as a key reaction. The availability of this advanced intermediate would facilitate the synthesis of Echinoside A and the related saponins from sea cucumbers.
AcknowledgmentThis work was financially supported by the Ministry of Science and Technology of China (No. 2013AA092903).
| [1] | (a) I. Kitagawa, T. Inamoto, M. Fuchida, et al., Structures of echinoside A and B, two antifugal oligoglycosides from the sea cucumber Actinopyga echinites (JAEGER), Chem. Pharm. Bull. 28 (1980) 1651-1653; (b) I. Kitagawa, M. Kobayashi, T. Inamoto, et al., Marine natural products. XIV. Structures of echinosides A and B, antifungal lanostane-oligosides from the sea cucumber Actinopyga echinites (JAEGER), Chem. Pharm. Bull. 33 (1985) 5214-5224; (c) M. Kobayashi, M. Hori, K. Kan, et al., Marine natural products. XXVII. Distribution of lanostane-type triterpene oligoglycosides in ten kinds of Okinawan sea cucumbers, Chem. Pharm. Bull. 39 (1991) 2282-2287. |
| [2] | M. Li, Z.H. Miao, Z. Chen, et al., Echinoside A, a new marine-derived anticancer saponin, targets topoisomerase2a by unique interference with its DNA binding and catalytic cycle, Ann. Oncol. 21 (2010) 597-607. |
| [3] | (a) Y. Yang, S. Laval, B. Yu, Chemical synthesis of saponins, Adv. Carbohydr. Chem. Biol. 37 (2014) 137-226; (b) B. Yu, J. Sun, Current synthesis of triterpene saponins, Chem. Asian J. 4 (2009) 642-654. |
| [4] | A. Bañó n-Caballero, G. Guillena, C. Ná jera, et al., Recoverable silica-gel supported binam-prolinamides as organocatalysts for the enantioselective solvent-free intra- and inter-molecular aldol reaction, Tetrahedron 69 (2013) 1307-1315. |
| [5] | A. Ali, C.F. Thompson, J.M. Balkovec, et al., Novel N-arylpyrazolo [3,2-c]-based ligands for the glucocorticoid receptor: receptor binding and in vivo activity, J. Med. Chem. 47 (2004) 2441-2452. |
| [6] | A.B. Smith, L. Kü rti, A.H. Davulcu, Y.S. Cho, Development of a scalable synthesis of a common eastern tricyclic lactone for construction of the nodulisporic acids, Org. Process Res. Dev. 11 (2007) 19-24. |
| [7] | H. Hisahiro, U. Hisashi, Optically pure (4aS)-(+)- or (4aR)-(-)-1,4a-dimethyl-4,4a, 7,8-tetrahydronaphthalene-2,5(3H,6H)-dione and its use in the synthesis of an inhibitor of steroid biosynthesis, J. Org. Chem. 53 (1988) 2308-2311. |
| [8] | (a) U. Eder, G. Sauer, R. Wiechert, New type of asymmetric cyclization to optically active steroid CD partial structures, Angew. Chem. Int. Ed. 10 (1971) 496-497; (b) P. Buchschacher, A. Fü rst, J. Gutzwiller, (S)-8a-Methyl-3,4,8,8a-tetrahydro-1,6 (2H, 7H)-naphthalenedione, Org. Synth. 63 (1985) 37. |
| [9] | (a) B. Bradshaw, E. Gorka, J. Bonjoch, et al., Efficient solvent-free Robinson annulation protocols for the highly enantioselective synthesis of the Wieland- Miescher ketone and analogues, Adv. Synth. Catal. 351 (2009) 2482; (b) G. Gabriela, N. Carmen, F.V. Santiago, N-Tosyl-(Sa)-binam-L-prolinamide as highly efficient bifunctional organocatalyst for the general enantioselective solvent- free Aldol reaction, Synlett 19 (2008) 3031. |
| [10] | D.N. Kirk, V. Petrow, Modified steroid hormones. Part XXVII. A new route to 4-methyl-3-oxo-Δ4-steroids, J. Chem. Soc. (1962) 1091. |
| [11] | P. Alexander, S. Karlheinz, Enantioselective total synthesis of (+)-labd-8(17)-ene- 3b, 15-diol and (-)-labd-8(17)-ene-3b,7a,15-triol, Tetrahedron Lett. 38 (1997) 2081-2084. |
| [12] | T.A. Spencer, R.A.J. Smith, D.L. Storm, R.M. Villarica, Total synthesis of (+-)-methyl vinhaticoate and (+)-methyl vouacapenate, J. Am. Chem. Soc. 93 (1971) 4856-4864. |
| [13] | A. Yajima, K. Mori, Diterpenoid total synthesis, XXXII synthesis and absolute configuration of (-)-phytocassane D, a diterpene phytoalexin isolated from the rice plant, Oryza sativa, Eur. J. Org. Chem. 65 (2000) 4079-4091. |