 2015, Vol.26
2015, Vol.26 


 , Hua Xieb , Wen-Hu Duana,d
, Hua Xieb , Wen-Hu Duana,d
b Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
c Pharmacology and Safety Evaluation Key Laboratory of Tibetan Medicine in Qinghai Province, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China;
d Department of Medicinal Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
Angiogenesis, the formation of new blood vessels from preexisting vessels, is a normal and vital process in growth and development, as well as in wound healing and female reproductive cycling [1, 2]. Pathological angiogenesis has been correlated with a variety of diseases, such as retinopathies, diabetes, rheumatoid arthritis, psoriasis and cancer [3, 4]. Tumor angiogenesis is a prerequisite for tumor growth as it is a fundamental step in the growth and metastasis of cancer [5]. This makes angiogenesis inhibition a promising therapeutic strategy against cancer.
Tumor angiogenesis is initiated by many factors, produced by both host and tumor cells, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) and other cytokines [6]. VEGFs and receptor (VEGFR-2) is very important in the direct regulation of angiogenesis, mitogenic signaling, and permeability-enhancing effects [7]. VEGFR-2 is expressed at high level in many kinds of cancers [8]. Disruption of VEGF signaling pathway by either specific binding of circulating VEGF or inhibiting receptor tyrosine kinases with small molecules has been found to inhibit angiogenesis, tumor progression and dissemination. Some VEGFR-2 inhibitors have been approved by FDA, such as Sunitinib [9], Sorafenib [10], Vandetanib [11], and Pazopanib [12] (Fig. 1).

|
Download:
|
| Fig. 1.Some reported VEGFR-2 inhibitors. | |
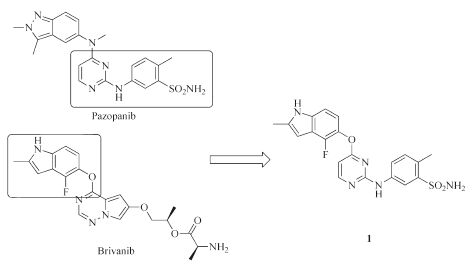
Brivanib (Fig. 2) was reported as an ATP competitive VEGFR-2 inhibitor with an IC50 of 25 nmol/L [13]. By analyzing the structure of Pazopanib and Brivanib, we speculated that the indole segment in Brivanib and indazole segment in Pazopanib may have similar interaction patterns with VEGFR-2. On the basis of this assumption, compound 1 (Fig. 2) was designed and synthesized. As expected, compound 1 potently inhibited VEGFR-2 with enzymatic IC50 values of 39 nmol/L. Encouraged by this promising result, we investigated the structure-activity relationship (SAR) of compound 1 by exploring changes on the pyrimidine core structure and the side chains. In this regard, a series of O-linked indole analogs were synthesized and tested in enzymatic level.

|
Download:
|
| Fig. 2.Design of hybrid compound 1. | |
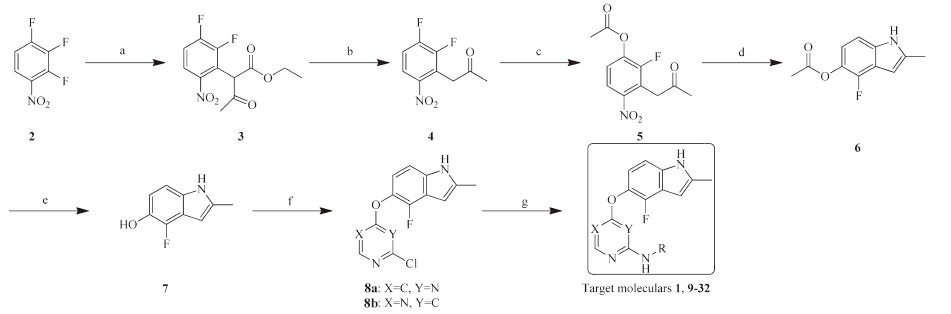
The synthesis of compounds 1, and 8-31 was shown in Scheme1. 2, 3, 4-Trifluornitrobenzene was chosen as starting material. Key intermediate (compound 7)was obtained according to the reported method [14]. 2, 4-Dicholoropyrimidine or 4, 6-dicholoropyrimidine was coupled with compound 7 to give 8a and 8b. All the analogs (1, 9-32) were prepared through an acid-catalyzed SNAr reaction of O-linked indole intermediates with substituted anilines. The structure of the new analogs was characterized by 1H NMR and MS. The detailed syntheses and characterizations are deposited in Supporting information. All analogs depicted in Table 1 were evaluated for their enzymatic activities against VEGFR-2.

|
Download:
|
| Scheme 1.Synthesis of compounds 1, and 9-32. Reagents and conditions: (a) NaH, ethyl acetoacetate, THF, 5 ℃ -r.t., 12 h; (b) HCl, HOAc, reflux, 12 h; (c) NaOAc, DMF, 100 ℃, 10 h; (d) H2, Pd/C, EtOH-DMF, 40 ℃, 10 h; (e) aq. NaHCO3, MeOH, r.t., 10 h; (f) 2,4-dicholoropyrimidine or 4,6-dicholoropyrimidine, aq. NaOH, acetone, 0-80 ℃; (g) substituted anilines, 36% HCl, i-PrOH, sealed tube, 100 ℃. | |
|
|
Table 1 VEGFR-2 inhibitory activity of compounds 1 and 9-32. |
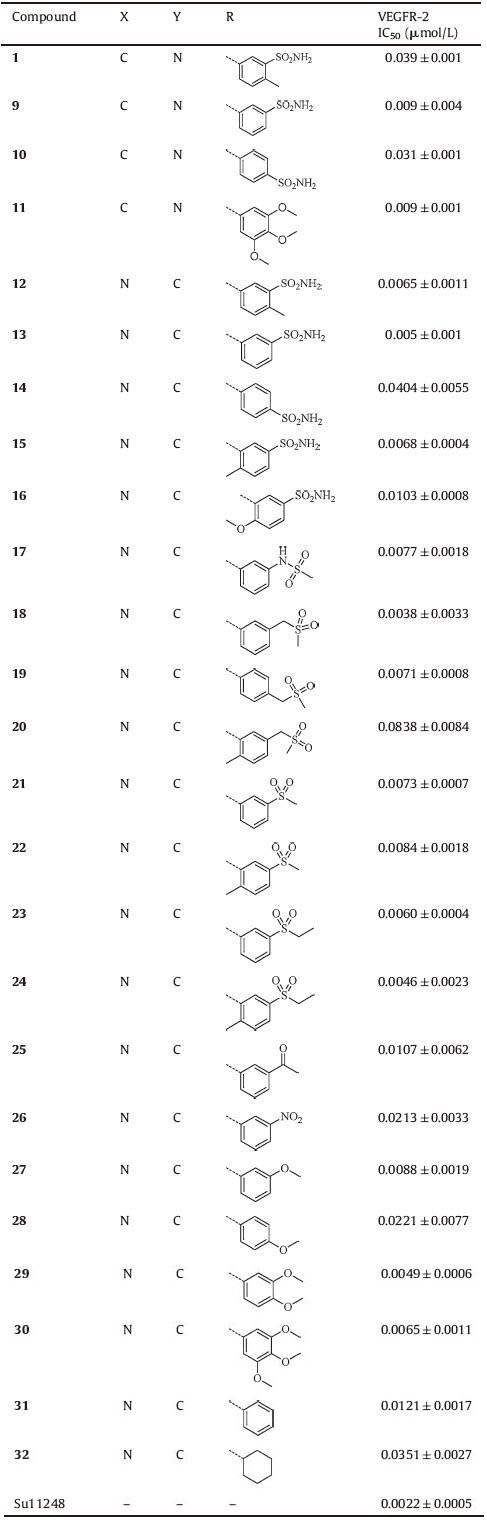
We firstly investigated the effect of substitution of the aniline at pyrimidine ring on the activity. Removal of methyl group led to compound 9, which exhibited higher potency on VEGFR-2 than compound 1 with an IC50 of 9 nmol/L. The result showed that the p-methyl group on the ring was not crucial to enzymatic activity. The para-sulfonamide analog (compound 10) showed a drastically decreased potency by 3.4-fold (IC50 = 31 nmol/L) than compound 9, which was indicated that the influence of the position of substituent was important. compound 11, with trimethoxy group on the ring, showed similar potency with compound 9 (IC50 = 31 nmol/L).
Next, 4, 6-disubstituted compound 12 was designed and synthesized. To our delight, this compound showed an improved VEGFR-2 inhibitory potency by 6-fold (IC50 = 6.5 nmol/L) than 2, 4-disubstituted compound 1. Then a series of 4, 6-substituted indole analogs were obtained as new scaffold for further research. Similarly, compound 13 with removal of p-methyl group increased activity by 1.3-fold (IC50 = 5 nmol/L) than compound 12. By variation of the position of substituent, compound 14 reduced potency with an IC50 of 40 nmol/L. Compounds 15 (IC50 = 6.8 nmol/L) and 16 (IC50 = 10.3 nmol/L) indicated that bulky group at the ortho-position of ring was not tolerable. Then a set of compounds (17-32) was obtained to investigate the effect of substitution pattern of the aniline at pyrimidine ring on the activity. As shown in Table 1, most of analogs showed good VEGFR-2 inhibitory activity. Analogs (compounds 17-26) with electron-withdrawing group displayed potent VEGFR-2 inhibitory activities. Among these analogs, compound 18 exhibited high inhibitory activitywith an IC50 value of 3.8 nmol/L. The meta-nitro substituted compound 26 exhibited weak activity (IC50 = 21.3 nmol/L) compared with other analogs. Then analogs (compounds 27-30) with methoxy group as electron donor were screened. compound 29 exhibited potent activity in enzymatic assay (IC50 = 4.9 nmol/L). compound 31, with no substituent on the ring also showed good activity with an IC50 value of 12.1 nmol/L. Interestingly, compound 32 with alkyl ring could inhibit VEGFR-2 with an IC50 value of 35.1 nmol/L. The SARs study revealed that this place can tolerate different substituent on the 4-position of pyrimidine.
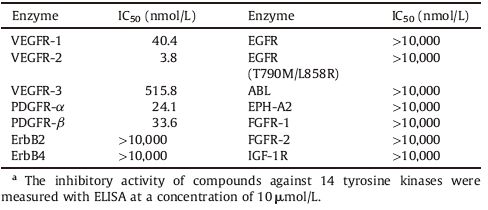
To assess the selectivity of this class of derivatives, compound 18 was further evaluated on a panel of tyrosine kinases. As shown in Table 2, compound 18 demonstrated good potency against VEGFR-1, PDGFR-α, and PDGFR-β, with IC50 values of 40.4, 24.1, and 33.6 nmol/L, respectively. However, it showed high selectivity over VEGFR-3 (135-fold, IC50 = 515.8 nmol/L) and excellent selectivity (>2530-fold) over ErbB2, ErbB4, EGFR, EGFR (T790M/L858R), ABL, EPH-A2, FGFR-1, FGFR-2 and IGF-1R.
|
|
Table 2 Kinase-selectivity profiling of compound 18.a |
{kind=link}
{kind=link}
{kind=link}
Finally, to further analyze the SAR of this series of analogs with O-linked indole scaffold, the docking model of compound 18 with VEGFR-2 was investigated (Fig. 3). Hydrogen bond was formed between the amide NH of indole with Glu885. Backbone amide NH and carbonyl oxygen of Cys919 formed hydrogen bonds with nitrogen of the pyrimidine and adjacent N-H of amine in the hinge region. The model study revealed the binding mode of VEGFR-2 to its inhibitor and helps to interpret the SAR of the O-linked indoles analogs.

|
Download:
|
| Fig. 3.Docking model of compound 18 with VEGFR-2 (PDB: 4ASD). | |
{kind=link}
In summary, a series of O-linked indoles analogs were designed and synthesized, and they were identified as potential VEGFR-2 inhibitors. Therefore, our results indicated that this class of compounds could be served as lead compound for the development of more selective anticancer medication.
AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. 81273365, 81173080 and 81321092), National Science & Technology Major Project ‘‘Key New Drug Creation and Manufacturing Program’’ (Nos. 2012ZX09103101-024 and 2014ZX09304002-008-001), Chinese National Programs for High Technology Research and Development (No. 2012AA020302) and the Shanghai Science and Technology Commission (No. 12DZ1930802) for their financial support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.cclet.2015.07.016.
| [1] | D.W. Siemann, D.J. Chaplin, M.R. Horsman, Vascular-targeting therapies for treatment of malignant disease, Cancer 100(2004) 2491-2499. |
| [2] | J. Folkman, M. Klagsbrun, Angiogenic factors, Science 235(1987) 442-447. |
| [3] | A. Levitzki, Protein kinase inhibitors as a therapeutic modality, Acc. Chem. Res. 36(2003) 462-469. |
| [4] | A. Garofalo, A. Farce, S. Ravez, et al., Synthesis and structure-activity relationships of (aryloxy)quinazoline ureas as novel, potent, and selective vascular endothelial growth factor receptor-2 inhibitors, J. Med. Chem. 55(2012) 1189-1204. |
| [5] | G. Gasparini, R. Longo, M. Toi, N. Ferrara, Angiogenic inhibitors:a new therapeutic strategy in oncology, Nat. Clin. Pract. Oncol. 2(2005) 562-577. |
| [6] | K. Sanphanya, S.K. Wattanapitayakul, S. Phowichit, V.V. Fokin, O. Vajragupta, Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach, Bioorg. Med. Chem. Lett. 23(2013) 2962-2967. |
| [7] | H.M.W. Verheul, H.M. Pinedo, Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition, Nat. Rev. Cancer 7(2007) 475-485. |
| [8] | Y. Takahashi, Y. Kitadai, C.D. Bucana, K.R. Cleary, L.M. Ellis, Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer, Cancer Res. 55(1995) 3964-3968. |
| [9] | H.K. Gan, B. Seruqa, J.J. Knox, Sunitinib in solid tumors, Expert Opin. Investig. Drugs 18(2009) 821-834. |
| [10] | G.M. Keating, A. Santoro, Sorafenib:a review of its use in advanced hepatocellular carcinoma, Drugs 69(2009) 223-240. |
| [11] | Y. Zhou, Y.L. Zhang, H.B. Zou, et al., The multi-targeted tyrosine kinase inhibitor vandetanib plays a bifunctional role in non-small cell lung cancer cells, Sci. Rep. 5(2015) 8629. |
| [12] | P.A. Harris, A. Boloor, M. Cheung, et al., Discovery of 5-[[4-[(2,3-dimethyl-2Hindazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor, J. Med. Chem. 51(2008) 4632-4640. |
| [13] | H. Huynh, V.C. Ngo, J. Fargnoli, et al., Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma, Clin. Cancer Res. 14(2008) 6146-6153. |
| [14] | R.S. Bhide, J.Y. Fan, P. Luca, et al., Process for preparing certain pyrrolotriazine compounds, WO2004009542. |