




Imidazo[1,2-a]pyridines are very important heterocycles which have been found to be the core scaffold of many natural products and drugs(Scheme1).Theyhavereceivedconsiderableinterest from the pharmaceutical industry because of their important biological activities and interesting therapeutic properties [1],including antibacterial [2a],antifungal [2b, 2c],antiviral [2d, 2f],antiulcer [2g],and anti-inflammatory behavior [2h]. Moreover,they can also be used in material science and molecular recognition [3].

|
Download:
|
| Scheme 1.The drugs containing imidazo[1,2-a]pyridine unit | |
By now,many synthetic methods for the construction of imidazo[1,2-a]pyridines have been reported. In general,the common strategies were the cyclocondensations of 2-aminopyridines with α-halocarbonyl compounds [4],1,3-dicarbonyl compounds [5],nitroolefins or alkynes [6]. Besides,the condensation of 2-aminopyridines,aldehydes and isonitriles or alkynes in one-pot was also an efficient method for the synthesis of imidazo[1,2- a]pyridines [7]. Some other novel synthetic approaches have been established in recent years. For example,the transition-metal catalyzed C-H activation or coupling methods have been demonstrated by some research groups recently [8]. Furthermore,several domino protocols have also been proposed for preparation of imidazo[1,2-a]pyridines,which avoided the requirement of any transition-metal [9, 10].
In our previous studies,we found that aryl methyl ketones could be quantitatively converted to aryloxoethanals in I2/DMSO system. The aryloxoethanals generated in situ were easily captured by 2-aminopyridines to afford 2,3-disubstituted imidazo[1,2- a]pyridines [10]. Based on these results,this work developed an I2/CuO-promoted one-pot protocol for the synthesis 2-substituted imidazo[1,2-a]pyridines via 2-aminopyridines capturing α-iodo acetophenones generated in situ from aryl methyl ketones in MeOH (Scheme 2).

|
Download:
|
| Scheme 2.Synthesis of imidazo[1,2-a]pyridines. | |
All aryl methyl ketones,β-ketone esters,2-aminopyridines,and other reagents were obtained from commercial suppliers and used without further purification. TLC analysis was performed using pre-coated glass plates. Column chromatography was performed using silica gel (200-300 mesh).
IR spectra were recorded on a Perkin-Elmer PE-983 infrared spectrometer as KBr pellets with absorption in cm-1. 1H NMR spectra were recorded on a Varian Mercury 400 or 600 MHz spectrometer. Chemical shifts are reported in ppm,relative to the internal standard of tetramethylsilane (TMS). HRMS were obtained on a Bruker Apex-Ultra 7.0T FTMS equipped with an electrospray source (ESI) or atmospheric-pressure chemical ionization (APCI). Melting points were determined using XT-4 apparatus and not corrected.
The synthetic route of the target products was outlined in Scheme 2. Finely powdered CuO (88 mg,1.1 mmol) and I2 (279 mg,1.1 mmol) were added to a solution of acetophenone 1a (120 mg,1 mmol) in anhydrous MeOH (20 mL). The mixture was refluxed for 1-10 h. When disappearance of the reactant (monitored by TLC),then added 2-aminopyridine 2a (94 mg,1.0 mmol) at reflux for another 2 h. After the reaction completed,the mixture was filtered and the solvent was removed under reduced pressure. The residue was poured into 10% Na2S2O3 solution (50 mL),the mixture was extracted with EtOAc (3 × 50 mL),and the organic layer was dried (Na2SO4). Removal of the solvent and purification of the residue by column chromatography gave the desired product 3aa as yellow solid 159.1 mg (yield 82%).
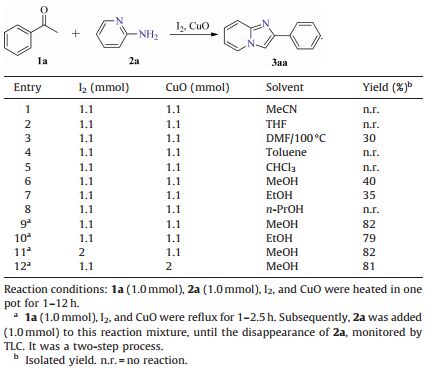
3. Results and discussionWe firstly selected acetophenone (1a) and 2-aminopyridine (2a) as model substrates to optimize the reaction conditions (Table 1). Several solvents were firstly examined. The reaction could not perform to give the desired product in acetonitrile,tetrahydrofuran,toluene,chloroform,and isopropanol (entries 1,2,4,5,and 8). However,the desired product was obtained in 30% yield when acetophenone (1a),2-aminopyridine (2a),I2 and CuO were heated in DMF at 100 ℃ for 1-2 h (entry 3). When the reaction was performed in methanol or ethanol,the target product could be obtained in 40% and 35% yields,respectively (entries 6 and 7). To our delight,the yields of the desired product were increased when acetophenone (1a),I2 and CuO were refluxed in methanol or ethanol for 1-2.5 h,with the subsequent addition of 2a for another 2 h (entries 9 and 10). Next,the equiv. of I2 and CuO was also optimized. Further elevating the amounts of I2 or CuO did not increase the yield obviously (entries 11 and 12). Above all,the optimal reaction condition turned out to be acetophenone 1a (1.0 mmol),I2 (1.1 mmol) and CuO (1.1 mmol) refluxed in MeOHfor 1-2.5 h,then 2-aminopyridine (2a) (1.0 mmol) was added at reflux for another 2 h.
| Table 1 Optimization of the reaction conditions. |
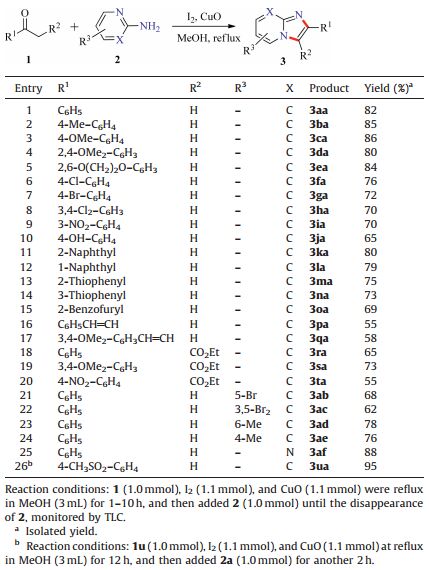
With suitable reaction conditions established,the generality of the reaction was explored (Table 2). The substrates with the electron-donating substituents on the benzene ring gave good yields,such as 4-Me,4-OMe,2,4-(OMe)2 and 2,6-O(CH2)2O groups (80%-86%,3ba-3ea). The electron-withdrawing substituent groups,such as 4-Cl,4-Br,3,4-Cl2,3-NO2 attached to the benzene ring shown a slight decrease in yields (70%-76%,3fa-3ia). It should be noted that the substrate with a sensitive hydroxy group (4-OH) in the phenyl group presented a moderate yield of 3ja (65%). Meanwhile,steric hindrance substrates 2-naphthyl methyl ketone and 1-naphthyl methyl ketone were also tolerant to this reaction to afford the desired products 3ka-3la in 79%-80% yields. Encouraged by the results obtained with aryl methyl ketones,we turned our attention to the heteroaryl ketones. The heterocycles,including thiophenyl and benzofuryl,did not affect the overall efficiency,and the corresponding products 3ma-3oa were furnished in 69%-75% yields. The unsaturated methyl ketones and β-ketone esters were also tested under the standard conditions. When unsaturated methyl ketones were selected as substrates,the corresponding products 3pa-3qa were obtained in 55%-58% yields. Moreover,β-ketone esters were also tolerant to the reaction to yield the corresponding products 3ra-3ta in moderate to good yields (55%-73%). Furthermore,the product 3fa was further determined by X-ray crystallographic analysis (Fig. 1).
| Table 2 The scope of ketones and 2-aminopyridines. |
{kind=link}
{kind=link}

|
Download:
|
| Fig. 1.The X-ray crystal structure of compound 3fa. | |
{kind=link}
To further expand the substrate scope,the substrates 2- aminopyridines were investigated. To our satisfaction,both electron-donating and electron-withdrawing groups attached to 2-aminopyridines were all suitable for this protocol,and provided the corresponding products (3ab-3ae) in 62%-78% yields. To our delight,2-aminopyrimidine was also tolerant to the reaction to afford the corresponding product 3af in 88% yield.
Zolimidine 3ua,which was the marketed antiulcer drug,could be synthesized in this concise route. As shown in Table 2,by utilizing 1-(4-(methylsulfonyl)phenyl)ethanone and 2-aminopyridine under I2/CuO-mediated conditions,the cyclization product 3ua could be generated smoothly in 95% yield.
To gain some insight into the mechanism of the reaction process,some control experiments were also performed (Scheme 3). It was found that acetophenone 1a could be converted into α-iodo ketone 1aa in 95% yield under I2/CuO-mediated conditions (Scheme 3a) [11a]. 1aa with 2-aminopyridine 2a could perform smoothly to afford product 3aa in 92% yield in MeOH at reflux conditions. Meanwhile,β-ketone ester 1r could also be transformed to intermediate 4r in 78% yield under I2/CuO-mediated conditions (Scheme 3b). Intermediate 4r could also react with 2a to furnish product 3ra in 90% yield in MeOH at reflux for 2 h.

|
Download:
|
| Scheme 3.The control experiments. | |
{kind=link}
Based on the above results,a possible mechanism of the present reaction was proposed as follows using acetophenone (1a) and 2-aminopyridine (2a) as an example (Scheme 4). Initially,the acetophenone 1a was converted to intermediate α-iodo acetophenone 1aa in the media of I2 and CuO [11]. The in situ generated intermediate 1aa underwent an intermolecular nucleophilic substitute with 2-aminopyridine 2a to obtain intermediate D,which was followed by isomerization to furnish intermediate E. Finally,intermediate E further underwent an intramolecular cyclization to afford the desired product 3aa. In the whole process,CuO plays multiple roles. It not only acts as the oxidizing agent or catalyst to convert molecular iodine into the reacting iodonium ion (I+) species,but also act as a weak base to neutralize hydrogen iodide,and reoxidize iodide ion (I-) into molecular iodine (I2),which forms together with insoluble copper(I) oxide and water [11a].

|
Download:
|
| Scheme 4.The plausible mechanism of the present reaction. | |
{kind=link}
In conclusion,an I2/CuO-promoted one-pot tandem strategy has been proposed for the synthesis of imidazo[1,2-a]pyridine derivatives. It is notable that the present reaction has a broad scope of substrates,including aryl methyl ketones,heteroaryl ketones,α,β-unsaturated methyl ketones,β-ketone esters,and substituted 2-aminopyridines. Owing to the generality of the reaction,this protocol should be of great utility in medical chemistry and organic methodology. Further investigations into the scope of this reaction and its applications are ongoing in our laboratory.
Acknowledgments
We would like to thank the National Natural Science Foundation of China (Nos. 21032001 and 21272085) for their generous financial support. We acknowlege the excellent doctorial dissertation cultivation grant from Central China Normal University Wuhan,China (No. 2013YBYB63). We also thank Dr. Chuanqi Zhou,Hebei University,for analytical support.
| [1] | A.R. Katritzky, Y.J. Xu, H. Tu, Regiospecific synthesis of 3-substituted imidazo[1,2- a]pyridines, imidazo[1,2-a]pyrimidines, and imidazo[1,2-c]pyrimidine, J. Org. Chem. 68 (2003) 4935–4937 (and references therein). |
| [2] | (a) Y. Rival, G. Grassy, G. Michel, Synthesis and antibacterial activity of some imidazo[1,2-a]pyrimidine derivatives, Chem. Pharm. Bull. 40 (1992) 1170–1176; (b) M.H. Fisher, A. Lusi, Imidazo[1,2-a]pyridine anthelmintic and antifungal agents, J. Med. Chem. 15 (1972) 982–985; (c) Y. Rival, G. Grassy, A. Taudou, R. Ecalle, Antifungal activity in vitro of some imidazo[1,2-a]pyrimidine derivatives, Eur. J. Med. Chem. 26 (1991) 13–18; (d) A. Gueiffier, M. Lhassani, A. Elhakmaoui, et al., Synthesis of acyclo-C-nucleosides in the imidazo[1,2-a]pyridine and pyrimidine series as antiviral agents, J. Med. Chem. 39 (1996) 2856–2859; (e) C. Hamdouchi, J. de Blas, M. del Prado, et al., 2-Amino-3-substituted-6-[(E)-1- phenyl-2-(N-methylcarbamoyl)vinyl]imidazo[1,2-a]pyridines as a novel class of inhibitors of human rhinovirus: stereospecific synthesis and antiviral activity, J. Med. Chem. 42 (1999) 50–59; (f) M. Lhassani, O. Chavignon, J.M. Chezal, et al., Synthesis and antiviral activity of imidazo[1,2-a]pyridines, Eur. J. Med. Chem. 34 (1999) 271–274; (g) J.J. Kaminsky, A.M. Doweyko, Antiulcer agents. 6. Analysis of the in vitro biochemical and in vivo gastric antisecretory activity of substituted imidazo[1,2- a]pyridines and related analogues using comparative molecular field analysis and hypothetical active site lattice methodologies, J. Med. Chem. 40 (1999) 427–436; (h) K.C. Rupert, J.R. Henry, J.H. Dodd, et al., Imidazopyrimidines, potent inhibitors of p38 MAP kinase, Bioorg. Med. Chem. Lett. 13 (2003) 347–350. |
| [3] | A.J. Stasyuk, M. Banasiewicz, M.K. Cyrański, D.T. Gryko, Imidazo[1,2-a]pyridines susceptible to excited state intramolecular proton transfer: one-pot synthesis via an Ortoleva–King reaction, J. Org. Chem. 77 (2012) 5552–5558. |
| [4] | (a) S. Ulloora, A.V. Adhikari, R. Shabaraya, Synthesis and antiepileptic studies of new imidazo[1,2-a]pyridine derivatives, Chin. Chem. Lett. 24 (2013) 853–856; (b) A. Herath, R. Dahl, N.D.P. Cosford, Fully automated continuous flow synthesis of highly functionalized imidazo[1,2-a]heterocycles, Org. Lett. 12 (2010) 412–415. |
| [5] | (a) X. Wang, L. Ma, W. Yu, Synthesis of imidazo[1,2-a]pyridines by the bis(acetyloxy)( phenyl)-λ3-iodane-mediated oxidative coupling of 2-aminopyridines with b-keto esters and 1,3-diones, Synthesis (2011) 2445–2453; (b) L.J. Ma, X.P. Wang, W. Yu, B. Han, TBAI-catalyzed oxidative coupling of aminopyridines with b-keto esters and 1,3-diones—synthesis of imidazo[1,2- a]pyridines, Chem. Commun. 47 (2011) 11333–11335. |
| [6] | (a) R.L. Yan, H. Han, C. Ma, et al., Cu(Ⅰ)-catalyzed synthesis of imidazo[1,2- a]pyridines from aminopyridines and nitroolefins using air as the oxidant, J. Org. Chem. 77 (2012) 2024–2028; (b) J. Zeng, Y.J. Tan, M.L. Leow, X.W. Liu, Copper(Ⅱ)/iron(Ⅲ) co-catalyzed intermolecular diamination of alkynes: facile synthesis of imidazopyridines, Org. Lett. 14 (2012) 4386–4389; (c) C.L. Zong, R.S. Zeng, J.P. Zou, One-pot copper nanoparticle-catalyzed synthesis of imidazo[1,2-a]pyridines under solvent-free conditions, Chem. J. Chin. Univ. 30 (2014) 632–638. |
| [7] | (a) S.K. Guchhait, A.L. Chandgude, CuSO4–glucose for in situ generation of controlled Cu(Ⅰ)–Cu(Ⅱ) bicatalysts: multicomponent reaction of heterocyclic azine and aldehyde with alkyne, and cycloisomerization toward synthesis of N-fused imidazoles, J. Org. Chem. 77 (2012) 4438–4444; (b) O.N. Burchak, L. Mugherli, M. Ostuni, J.J. Lacapère, M.Y. Balakirev, Combinatorial discovery of fluorescent pharmacophores by multicomponent reactions in droplet arrays, J. Am. Chem. Soc. 133 (2011) 10058–10061. |
| [8] | (a) Q. Liao, L.Y. Zhang, S.T. Li, C.J. Xi, Domino N–H/C–H bond activation: coppercatalyzed synthesis of nitrogen-bridgehead heterocycles using azoles and 1,4-dihalo-1,3-dienes, Org. Lett. 13 (2011) 228–231; (b) H. Wang, Y. Wang, C. Peng, J. Zhang, Q. Zhu, A direct intramolecular C–H amination reaction cocatalyzed by copper(Ⅱ) and iron(Ⅲ) as part of an efficient route for the synthesis of pyrido[1,2-a]benzimidazoles from N-aryl-2-aminopyridines, J. Am. Chem. Soc. 132 (2010) 13217–13219. |
| [9] | N. Shao, G.X. Pang, C.X. Yan, G.F. Shi, Y. Cheng, Reaction of β-lactam carbenes with 2-pyridyl isonitriles: a one-pot synthesis of 2-carbonyl-3-(pyridylamino)imidazo[ 1,2-a]pyridines useful as fluorescent probes for mercury ion, J. Org. Chem. 76 (2011) 7458–7465. |
| [10] | Z. Fei, Y.P. Zhu, M.C. Liu, F.C. Jia, A.X. Wu, I2-promoted direct one-pot synthesis of 2-aryl-3-(pyridine-2-ylamino)imidazo[1,2-a]pyridines from aromatic ketones and 2-aminopyridines, Tetrahedron Lett. 54 (2013) 1222–1226. |
| [11] | (a) G.D. Yin, M. Gao, N.F. She, et al., Highly efficient and clean method for direct aiodination of aromatic ketones, Synthesis (2007) 3113–3117; (b) Z.H. Wang, G.D. Yin, J. Qin, et al., An efficient method for the selective iodination of a,b-unsaturated ketones, Synthesis (2008) 3675–3681; (c) G.D. Yin, B.H. Zhou, X.G. Meng, A.X. Wu, Y.J. Pan, Efficient C–C double-bond formation reaction via a new synthetic strategy: a self-sorting tandem reaction, Org. Lett. 8 (2006) 2245–2248; (d) M. Gao, G.D. Yin, Z.H. Wang, et al., A concise and efficient way to synthesize polyenic diones directly from a,β-unsaturated methyl ketones, Tetrahedron 65 (2009) 6047–6049. |