




b State Key Laboratory of Medicinal Chemical Biology, College of Pharmacy, Nankai University, Tianjin 300071, China
Inositol phosphates (IPs),as important second messengers of signal transduction,regulate many biological functions in cellular signaling [1, 2, 3, 4, 5],which are related to various physical actions such as cell proliferation,muscle contraction,apoptosis,and gene transcription [6]. IPs also regulate DNA editing and repair [7], vesicle transport [8],and ion channel regulation [9],and thus have been implicated in diseases such as cancer and diabetes [10]. Among the known IPs,D-myo-inositol 1,4,5-trisphosphate (InsP3, IP3),which is generated by the intracellular phosphatidylinositolspecific phospholipase C (PI-PLC)-mediated hydrolysis of phosphatidylinositol 4,5-bisphosphate (PtdInsP2),is a biologically important intracellular Ca2+ -mobilizing second messenger. InsP3 interacts specifically with its receptors (InsP3R) on the endoplasmic or sarcoplasmic reticulum membrane to release intracellular Ca2+ into the cytosol [11, 12, 13, 14, 15].
The biochemical importance of InsP3 has promoted many syntheses of InsP3 and their derivatives. Although a lot of active analogues attained similar potencies to that of InsP3,little has surpassed it [16]. The only breakthrough in this area was the discovery of adenophostins A and B,isolated from culture broths of Penicillium brevicompactumin 1993 [17] and were regarded as functionally analogous to InsP3. With 10- to 100-fold greater potencies for receptor affinity [18, 19, 20, 21] and Ca2+ release [22] than InsP3 itself,adenophostins A and B opened up a whole new chapter in searching for antagonists or agonists of InsP3 receptors (InsP3R). However,the difficulties in synthesizing these InsP3-like derivatives hindered the step of using these compounds as probes to investigate intracellular Ca2+ signal transduction and its mechanism. The other main obstacle is the instability of inositol phosphate analogues to kinase,phosphatases,and lipases,which impedes further development of potentially beneficial drugs [23].
Although the generation,mobilization,release of Ca2+ and transduction,termination of signal mediated by inositol phosphates are well known to date through several decades of intense studies by biologists and chemists,this area is far more complex and difficult than that originally imagined as investigation became more in-depth. It is therefore clear that the development of selective,membrane-permeant and enzymatic stable InsP3R analogues is vital to enable further studies of Ca2+ and InsP3 signaling.
Phosphonate,an isostere of phosphate,was extensively used to replace phosphate and acted as phosphorylated substrates [24] because of its stability toward the action of phosphatases [25, 26] and PI-specific phospholipases C [27]. Moreover,some inositol phosphonate analogues with the core structure ‘‘inositol-CH2-P’’ have been synthesized to study their biological functions [28, 29, 30, 31, 32]. Studies with several cancer cell lines have shown that signal transduction mediated by inositol phosphates and phosphatidylinositol phosphates potentiated the effects of various cancer cell lines,which suggested that they could be employed in combination drug therapy [33, 34, 35]. There are also other examples [36, 37, 38, 39] that phosphonates replaced phosphates in the drug development. We previously reported a kind of inositol phosphonate analogues usingmyo-inositol as the starting material,and found that two phosphonate analogues exhibited relatively good cytotoxicity against non-small cell lung cancer (NSCLC) cell line A549 [40].
Therefore,phosphonate moiety instead of the phosphate ester group is a good alternative to resistant to enzymatic degradation and has already achieved great success in the development of antivirus drugs [39]. In continuation of our research program on pursuing novel biologically active compounds in the fields of human health,especially anticancer agents,we reported herein the design and synthesis of a series of inositol phosphonate analogues with the general structure ‘‘inositol-O-CH2-P’’ or ‘‘inositol-O- CH2CH2-P’’ along with the initial antitumor activity evaluation results. 2. Experimental
Solvents were obtained from commercial sources and purified according to the literature [41] if necessary. Nuclear magnetic resonance spectra were recorded on Bruker AV 300 or 400 NMR instrument in CDCl3. Chemical shifts (d) are reported in parts per million (ppm),relative to TMS as internal standard. High resolution mass spectra (HR-MS) were carried out on IonSpec FTICR-MS instrument from Varian using ESI as ionization mode. Flashcolumn chromatography was performed using commercial grades of silica gel 200-300 meshes. Analytical thin layer chromatography (TLC) was performed on pre-coated silica gel 60 F254 plates,and spot visualization was accomplished by UV light (254 nm) or phosphomolybdic acid solution. 2.1. General procedure for synthesis of methylenephosphonate
inositol compounds 3aa-3adand3ba-3bd To a stirred solution of mono-hydroxy inositol 2a-d (0.25 mmol) in dry THF (5 mL) at-60℃ under N2was addednbutyllithium (2.5 mol/L in THF,0.3 mmol) by syringe. After string 30 min,the resulting mixture was added to (dialkoxyphosphinyl) methyl triflate 1a or 1b(0.325 mmol) in 1 mL of dry THF by syringe. Then the reactant was allowed to warm up to 0℃overa period of 1 h and stirred for 1 h at 0℃. After the completion of starting material by thin layer chromatography,the reactant was quenched by saturated NH4Cl aqueous (5 mL),and extracted with DCM (15 mL×4),dried over Na2SO4. After concentration of DCM,the residue was purified by chromatography on silica gel (PE/EA = 2:1-1:1) to give compound 3 as an oil. 2.2. General procedure for synthesis of ethylenephosphonate inositol compounds 5a-d
A mixture of bromoethyl inositol 4a-d(0.03 mmol)and triethyl phosphite (3 mL) was heated to 140℃ for 10 h,TLC showed the starting material was consumed completely. Then the mixture was concentrated and the residue was separated by chromatography on silica gel (PE/EA = 3:1-1:1) to give compound5as an oil. 2.3. Bioassay
NSCLC cell line A549 was obtained from ATCC (American type culture collection) and were maintained in 5% CO2at 37℃. A549 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS,Omega Scientific) and 1% penicillin/streptomycin (Omega Scientific). Approximately 1000 cells were seeded into individual wells of 96-well tissue culture plates and incubated for 12 h,medium was 0.2 μL/well. The compound was diluted to 10μg/mL using DMEM (final concentration in the well),to analyze the inhibition effect on A549 roughly. The positive inhibitors were dissolved in DMSO reaching a final DMSO concentration of 0.5%. Viability was normalized to control cells which were treated with the vehicle, DMSO. After 72 h incubation at 37℃ and 5% CO2,cell viability was assessed by MTT assay. Cells were replenished with fresh medium (0.1 μL/well) which contains 10% MTT. Culture medium was removed after 4 h,and the formazan was dissolved in DMSO (200μL/well). Then OD570 were measured by Plate Reader (BIORAD). 3. Results and discussion
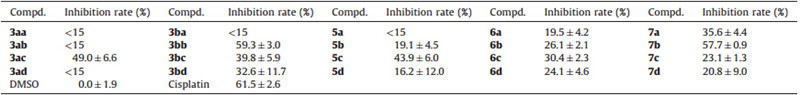
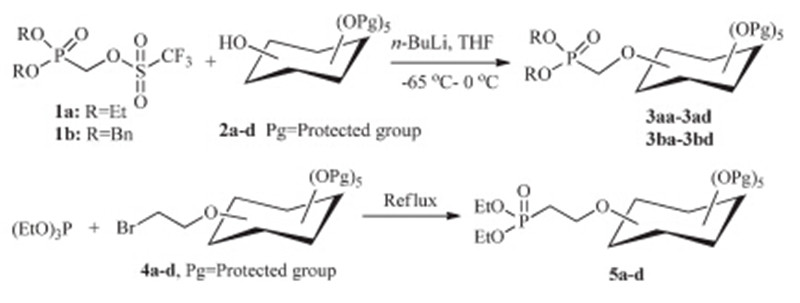
In our previous work,we found mono-phosphate inositol compounds had low anticancer activity (see Table 1) against nonsmall cell lung cancer cell. We thus postulated that the instability of the phosphates to most enzymes in the cell may compromise their activities. Based on this hypothesis,we designed and synthesized the phosphonate inositol derivatives as alternatives of phosphate inositol compounds by introducing the known structural features such as P-CH2-O and P-CH2CH2-O fragments at the different positions of the inositol ring. In fact,the use of phosphonates or phosphonic acids as analogues of natural phosphates represents a more systematic approach to metabolic regulation and enhancement or inhibition,than is commonly attributed to ‘‘analogue’’ study [39]. The synthetic strategies of these novel phosphonate inositol derivatives were depicted in Scheme 1. A nucleophilic substitution of trifluoromethanesulfonate-activated phosphonates with the protected inositols afforded compounds 3 containing P-CH2-O fragment,whereas an Arbuzov rearrangement of triethylphosphite with bromoethylinositols furnished compounds5 with P-CH2CH2-O fragment.
| Table 1 The inhibition rate against A549 (10mg/mL). |

|
Download:
|
| Scheme 1.Synthetic route of titled phosphonate inositol compounds 3and5. | |
{kind=link}
The starting (dialkyloxyphosphinyl)methyltriflate 1a and 1b were prepared according to the reported procedures in three steps from PCl3[42, 43, 44]. Reaction of paraformaldehyde with diethylphosphite or dibenzylphosphite gave the dialkyl (hydroxymethyl)phosphonate,which was converted to triflate 1a,1b using 2,6-lutidine as the base in total yield of 38% and 30%,respectively (Scheme 2).

|
Download:
|
| Scheme 2.Synthesis of (dialkyloxyphosphinyl)methyltriflate1aand 1b. | |
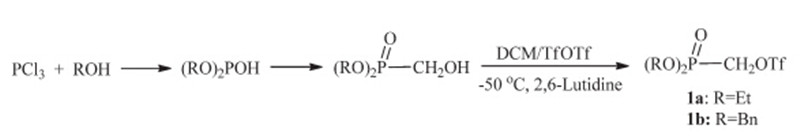{kind=link}
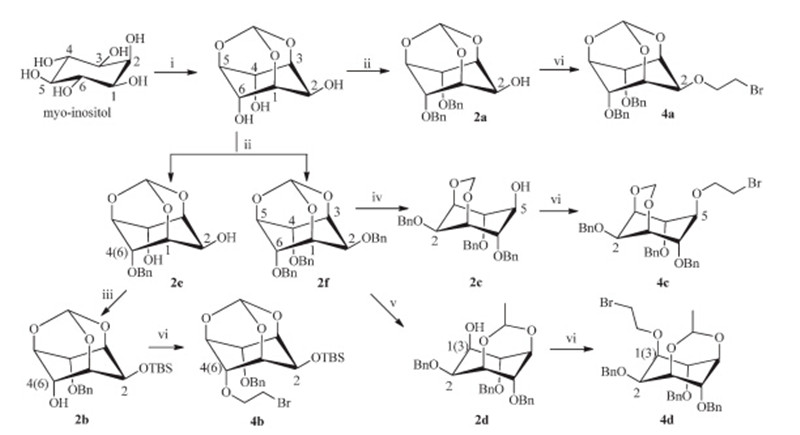
Mono-hydroxyl inositol compounds 2 were obtained by selective protection or ring-opening from commercially available myo-inositol in two to three steps with desired yields [23]. Bromoethyl inositol compounds 4a-d were generated from the mono-hydroxyl inositol compounds with 2-bromoethyl trifluoromethanesulfonate using 2,6-lutidine as the base. Although we could not achieve satisfactory results by direct substitution of protected inositol 2a-d with dibromoethane in basic condition such as NaH,n-BuLi or DBU due to the low reactivity of the reactants,we used the more reactive 2-bromoethyl trifluoromethanesulfonate instead of dibromoethane to make the desired bromoethyl inositol 4a-d under lower temperature in acceptable yields (Scheme 3).

|
Download:
|
| Scheme 3.Synthesis of mono-hydroxyl inositol compounds 2a–d and bromoethyl inositol4a–d. Reagents and conditions: (i) HC(OEt)3, PTSA monohydrate, DMF, 110 ℃, 28 h, 83%; (ii) NaH, BnBr, DMF, 0℃–r.t., overnight, 68% for 2a, 48% for 2e, 74% for 2f; (iii) TBDMSCl, imidazole, DMF, r.t., overnight, 90%; (iv) DIBAL, CH2Cl2,0℃–r.t., 4 h, 95%; (v) AlMe3,CH2Cl2,0℃–r.t., 5 h, 68%; (vi) BrCH2CH2OTf, THF,-60–0℃, 2 h, 20% for4a, 44% for 4b, 32% for 4c, 40% for 4d. | |
{kind=link}
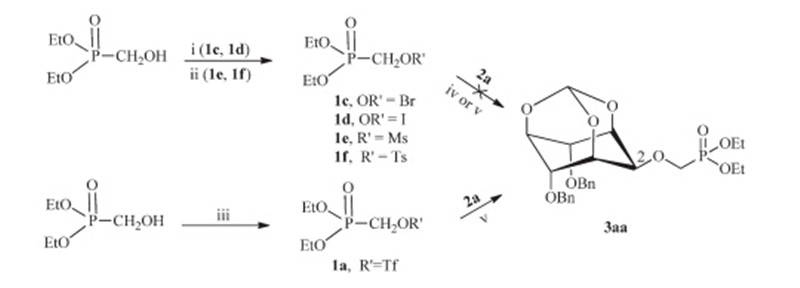
After protected mono-hydroxyl inositol derivatives 2a-d,our first attempt to synthesize the final compounds 3aa used (RO)2P(O)CH2Br,(RO)2P(O)CH2I,(RO)2P(O)CH2OTs or (RO)2P(O)-CH2OMs as the phosphorylation reagents to react with 2a under basic condition (NaH orn-BuLi),but no expected product was obtained (Scheme 4). The reason may be the less reactive reactants because of the steric hindrance of the protected inositol and the weak electrophilicity of the phosphorylation reagents. So the effective method was to enhance the reactivity of the phosphorylation reagent or the protected inositol. Trifluoromethanesulfonate (triflate) is an excellent leaving group and has been widely used in organic synthesis [45]. Meanwhile (dialkyloxyphosphinyl)methyltriflate is also easy to be obtained [46]. We therefore synthesized the (dialkyloxyphosphinyl)methyltriflate 1a and 1b, which were then treated with nucleophiles 2a-d under basic condition to afford the title compounds 3 in good yields.

|
Download:
|
| Scheme 4.Synthesis of methylenephosphonateinositol compounds 3aa. Reagents and conditions: (i) TsCl, TEA, DCM; NaI or NaBr, acetone, reflux; (ii) TsCl or MsCl, TEA, DCM; (iii) Tf2O, 2,6-lutidine, DCM,-50℃, 71%; (iv) NaH, DMF, 0℃–r.t., overnight; and (v)n-BuLi, THF,-65–0℃. | |
{kind=link}
With respect to compound5,we attempted to prepare them according to the procedure of compounds3with 2-(diethoxyphosphinyl)ethyltriflate ((EtO)2P(O)CH2CH2OTf) instead of (diethoxyphosphinyl)methyl triflate 1a,but were not successful. The reason of the unsuccessful attempts could be the b-elimination reaction of (EtO)2P(O)CH2CH2OTf under basic condition to give the a,b-unsaturated phosphonate,diethyl vinylphosphonate,whose structure was determined by 1H NMR and 31P NMR. 1H NMR (400 MHz,CDCl3):δ6.27-5.94 (m,3H,CH255CH),4.03-3.99 (m,4H, 2×CH3CH2O),1.25 (t,6H,J= 7.0 Hz,2×CH3CH2O); 13CNMR (101 MHz,CDCl3):δ135.36,125.89 (d,J= 184.83 Hz),61.76 (d, J= 5.05 Hz),16.25 (d,J= 6.06 Hz); 31P NMR (162 MHz,CDCl3):δ 17.18. Numerous methods have been reported in the literature for the synthesis of phosphonate besides the above nucleophilic substitution,the best known of which are the Arbuzov and Michaelis-Becker reactions [47]. Because of theb-elimination reaction of (EtO)2P(O)CH2CH2OTf,we thus tried the Arbuzov rearrangement to prepare the title compounds 5. As shown in Scheme 1,the mixture of triethylphosphite and brominated inositol compounds 4a-d was refluxed for 10 h to obtain the final products in good yields.
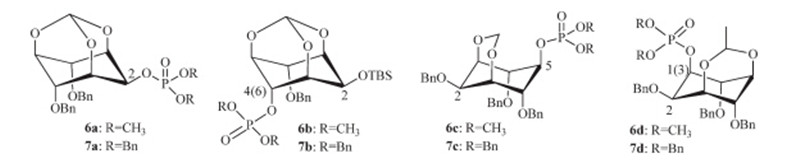
Having obtained the inositol mono-phosphonates,we then assessed the antitumor activity against non-small cell lung cancer (NSCLC) cell line A549. The results show most compounds have low inhibition against A549,except compounds 3acand 3bb, which have about 50%-60% inhibition against A549,similar to that of the positive control,Cisplatin. The structure-activity relationship (SAR) shows benzyl phosphonates 3bb and3bdexhibit more potential anticancer activity than ethyl phosphonates 3ab and3ad. However,adding one methylene between inositol ring and phosphorus atom (compounds5) seems no improvement on the anticancer activity in comparison with compounds3aa-3ad. The phosphorylation at the 2-position on inositol is not favorable (3aa, 3ba,5a),while other positions have little effect on the anticancer activity. In our previous work,we obtained some phosphate inositols 6and 7,and the anticancer activity results indicate compound3ac,3bc and3bdhave significantly activity improvement in comparison with phosphate inositol6c,7c and 7d. The compounds 3bb and7b have the best activity,which indicate the tert-butyldimethylsilyl (TBS) group at the 2-position on inositol in compounds 3 and7 is favorable for their anticancer activities (Scheme 5).

|
Download:
|
| Scheme 5.The structures of compounds 6 and7 | |
{kind=link}
In summary,we prepared 12 mono-phosphonate inositol compounds by two kinds of strategies,i.e.,nucleophilic substitution and Arbuzov rearrangement,respectively. The structures of these novel inositol phosphonates were confirmed by NMR and HRMS. Among them,two phosphonate analogues (3ac and 3bb) exhibited good antitumor activity against cell line A549 and the preliminary SAR was discussed. Further study to optimize the model of these analogues is being actively pursued. The variable structure and unique property of this type of compounds make them good mechanistic chemical probes and useful drug-like leads.
AcknowledgmentsThis work was financially supported by Tianjin Municipal Natural Science Foundation (Key Program No. 12JCZDJC22000) and Ministry of Science and Technology of China (Nos. 2010CB126102, 2011BAE06B05).
| [1] | M. Bennett, S.M.N. Onnebo, C. Azevedo, et al., Inositol pyrophosphates: metabolism and signaling, Cell. Mol. Life Sci. 63 (2006) 552-564. |
| [2] | R.F. Irvine, M.J. Schell, Back in the water: the return of the inositol phosphates, Nat. Rev. Mol. Cell Biol. 2 (2001) 327-338. |
| [3] | V. Gosein, G.J. Miller, Roles of phosphate recognition in inositol 1,3,4,5,6-pentakisphosphate 2-kinase (IPK1) substrate binding and activation, J. Biol. Chem. 288 (2013) 26908-26913. |
| [4] | F.S. Menniti, K.G. Oliver, J.W. Putney Jr., et al., Inositol phosphates and cell signaling: new views of InsP5 and InsP6, Trends Biochem. Sci. 18 (1993) 53-56. |
| [5] | J.D. York, Regulation of nuclear processes by inositol polyphosphates, Biochim. Biophys. Acta 1761 (2006) 552-559. |
| [6] | M.J. Berridge, M.D. Bootman, H.L. Roderick, Calcium signalling: dynamics, homeostasis and remodelling, Nat. Rev. Mol. Cell Biol. 4 (2003) 517-529. |
| [7] | L.A. Hanakahi, M. Bartlet-Jones, C. Chappell, et al., Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair, Cell 102 (2000) 721-729. |
| [8] | J.M. Hilton, M. Plomann, B. Ritter, et al., Phosphorylation of a synaptic vesicleassociated protein by an inositol hexakisphosphate-regulated protein kinase, J. Biol. Chem. 276 (2001) 16341-16347. |
| [9] | M. Vajanaphanich, C. Schultz, M.T. Rudolf, et al., Long-term uncoupling of chloride secretion from intracellular calcium levels by Ins(3,4,5,6)P-4, Nature 371 (1994) 711-714. |
| [10] | Y. Shi, A.N. Azab, M.N. Thompson, et al., Inositol phosphates and phosphoinositides in health and disease, in: A.L. Majumder, B.B. Biswas (Eds.), Subcellular Biochemistry, 2006, 265-292. |
| [11] | M.J. Berridge, P. Lipp, M.D. Bootman, The versatility and universality of calcium signalling, Nat. Rev. Mol. Cell Biol. 1 (2000) 11-21. |
| [12] | R.F. Irvine, 20 years of Ins(1,4,5)P-3, and 40 years before, Nat. Rev. Mol. Cell Biol. 4 (2003) 586-590. |
| [13] | M.J. Berridge, R.F. Irvine, Inositol trisphosphate, a novel 2nd messenger in cellular signal transduction, Nature 312 (1984) 315-321. |
| [14] | K.M. Sureshan, A.M. Riley, A.M. Rossi, et al., Activation of IP3 receptors by synthetic bisphosphate ligands, Chem. Commun. (2009) 1204-1206. |
| [15] | M.J. Berridge, Inositol trisphosphate and calcium signaling, Nature 361 (1993) 315-325. |
| [16] | A.M. Riley, A.J. Laude, C.W. Taylor, et al., Dimers of D-myo-inositol 1,4,5-trisphosphate: design, synthesis, and interaction with Ins(1,4,5)P-3 receptors, Bioconjugate Chem. 15 (2004) 278-289. |
| [17] | M. Takahashi, T. Kagasaki, T. Hosoya, et al., Adenophostin-a and adenophostin-b -potent agonists of inositol-1,4,5-trisphosphate receptor produced by penicilliumbrevicompactum -taxonomy, fermentation, isolation, physicochemical and biological properties, J. Antibiot. 46 (1993) 1643-1647. |
| [18] | C.E. Adkins, F. Wissing, B.V.L. Potter, et al., Rapid activation and partial inactivation of inositol trisphosphate receptors by adenophostin A, Biochem. J. 352 (2000) 929-933. |
| [19] | V. Correa, A.M. Riley, S. Shuto, et al., Structural determinants of adenophostin A activity at inositol trisphosphate receptors, Mol. Pharmacol. 59 (2001) 1206-1215. |
| [20] | S. Shuto, K. Tatani, Y. Ueno, et al., Synthesis of adenophostin analogues lacking the adenine moiety as novel potent IP3 receptor ligands: some structural requirements for the significant activity of adenophostin A, J. Org. Chem. 63 (1998) 8815-8824. |
| [21] | J. Hirota, T. Michikawa, A. Miyawaki, et al., Adenophostin-medicated quantal Ca2+ release in the purified and reconstituted inositol 1,4,5-trisphosphate receptortype-1, FEBS Lett. 368 (1995) 248-252. |
| [22] | M. Takahashi, K. Tanzawa, S. Takahashi, Adenophostins, newly discovered metabolites of penicillium-brevicompactum, act as potent agonists of the inositol 1,4,5-trisphosphate receptor, J. Biol. Chem. 269 (1994) 369-372. |
| [23] | F. Song, J. Zhang, Y. Zhao, et al., Synthesis and antitumor activity of inositol phosphotriester analogues, Org. Biomol. Chem. 10 (2012) 3642-3654. |
| [24] | R. Engel, Phosphonates as analogues of natural phosphates, Chem. Rev. 77 (1977) 349-367. |
| [25] | A.J. Ganzhorn, J. Hoflack, P.D. Pelton, et al., Inhibition of myo-inositol monophosphatase isoforms by aromatic phosphonates, Bioorg. Med. Chem. 6 (1998) 1865-1874. |
| [26] | W. Huang, H. Zhang, F. Davrazou, et al., Stabilized phosphatidylinositol-5-phosphate analogues as ligands for the nuclear protein ING2: chemistry, biology, and molecular modeling, J. Am. Chem. Soc. 129 (2007) 6498-6506. |
| [27] | M.S. Shashidhar, J.F. Keana, J.J. Volwerk, et al., Synthesis of phosphonate derivatives of myo-inositol for use in biochemical studies of inositol-binding proteins, Chem. Phys. Lipids 53 (1990) 103-113. |
| [28] | J.R. Falck, A. Abdali, S.J. Wittenberger, Total synthesis of the 5-methylenephosphonate analog of D-myo-inositol 1,4,5-trisphosphate, J. Chem. Soc. Chem. Commun. (1990) 953-955. |
| [29] | N.S. Keddie, Y.L. Ye, T. Aslam, et al., Development of inositol-based antagonists for the D-myo-inositol 1,4,5-trisphosphate receptor, Chem. Commun. 47 (2011) 242-244. |
| [30] | D. Vizitiu, A.G. Kriste, A.S. Campbell, et al., Inhibition of phosphatidylinositolspecific phospholipase C: studies on synthetic substrates, inhibitors and a synthetic enzyme, J. Mol. Recognit. 9 (1996) 197-209. |
| [31] | M.S. Shashidhar, J.J. Volwerk, J.F. Keana, et al., Inhibition of phosphatidylinositolspecific phospholipase C by phosphonate substrate analogues, Biochim. Biophys. Acta 1042 (1990) 410-412. |
| [32] | Y. Wu, C. Zhou, M.F. Roberts, Stereocontrolled syntheses of water-soluble inhibitors of phosphatidylinositol-specific phospholipase C: inhibition enhanced by an interface, Biochemistry 36 (1997) 356-363. |
| [33] | W. Xie, H.R. Peng, D.I. Kim, et al., Structure-activity relationship of aza-steroids as PI-PLC inhibitors, Bioorg. Med. Chem. 9 (2001) 1073-1083. |
| [34] | M. Ryan, M.P. Smith, T.K. Vinod, et al., Synthesis, structure-activity relationships, and the effect of polyethylene glycol on inhibitors of phosphatidylinositol-specific phospholipase C from Bacillus cereus, J. Med. Chem. 39 (1996) 4366-4376. |
| [35] | W.G. Xie, H.R. Peng, L.H. Zalkow, et al., 3β-Hydroxy-6-aza-cholestane and related analogues as phosphatidylinositol specific phospholipase C (PI-PLC) inhibitors with antitumor activity, Bioorg. Med. Chem. 8 (2000) 699-706. |
| [36] | P. Nasomjai, D. O’Hagan, A.M.Z. Slawin, Synthesis of phosphonate and phostone analogues of ribose-1-phosphates, Beilstein J. Org. Chem. 5 (2009). |
| [37] | W.B. Wan, J.R. Beadle, C. Hartline, et al., Comparison of the antiviral activities of alkoxyalkyl and alkyl esters of cidofovir against human and murine cytomegalovirus replication in vitro, Antimicrob. Agents Chemother. 49(2005) 656-662. |
| [38] | I.E. Glowacka, J. Balzarini, A.E. Wroblewski, Synthesis and biological evaluation of novel 1,2,3-triazolonucleotides, Arch. Pharm. 346 (2013) 278-291. |
| [39] | E. De Clercq, A. Holy, Acyclic nucleoside phosphonates: a key class of antiviral drugs, Nat. Rev. Drug Discov. 4 (2005) 928-940. |
| [40] | F. Song, J. Zhang, Q. Cui, et al., Synthesis and antitumour activity of inositol phosphonate analogues, Tetrahedron Lett. 53 (2012) 1102-1104. |
| [41] | W.L.F. Armarego, C.L.L. Chai, Purification of Laboratory Chemicals, fifth ed., Butterworth-Heinemann, Burlington, MA, USA, 2003. |
| [42] | A.F. Kluge, Diethyl [(2-tetrahydropyranyloxy)methyl] phosphonate, Org. Synth. 7(1990) 160-161. |
| [43] | H. Ikeda, E. Abushanab, V.E. Marquez, The assembly of beta-methylene-TAD, a metabolically stable analogue of the antitumor agent TAD, by the stepwise esterification of monodeprotected methylenebis(phosphonate) benzyl esters under Mitsunobu conditions, Bioorg. Med. Chem. Lett. 9 (1999) 3069-3074. |
| [44] | Y.B. Xu, M.T. Flavin, Z.Q. Xu, Preparation of new Wittig reagents and their application to the synthesis of a,β-unsaturated phosphonates, J. Org. Chem.61 (1996) 7697-7701. |
| [45] | P.J. Stang, M. Hanack, L.R. Subramanian, Perfluoroalkanesulfonic esters: methods of preparation and applications in organic chemistry, Synthesis (1982) 85-126. |
| [46] | D.P. Phillion, S.S. Andrew, Synthesis and reactivity of diethyl phosphonomethyltriflate, Tetrahedron Lett. 27 (1986) 1477-1480. |
| [47] | A.K. Bhattacharya, G. Thyagarajan, Michaelis-Arbuzov rearrangement, Chem. Rev. 81 (1981) |