




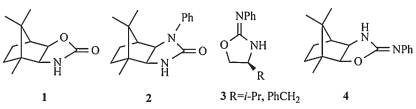
Chiral auxiliary-derived asymmetric alkylations have been studied extensively and are now important and useful methods for constructing synthetic building blocks of high stereoisomeric purity [1, 2, 3, 4, 5, 6, 7]. The need for such building blocks has inspired many researchers to develop a large number of chiral auxiliaries for the induction of asymmetry in a wide variety of reactions [8, 9, 10, 11]. Recently there has been considerable interest in the development of camphor-based new chiral auxiliaries from both synthetic and mechanistic points of view [12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22]. Camphor-based auxiliaries, such as1[16] and2[15] (Fig. 1),were known to have excellent diastereoselectivity when the sterically congested camphor moiety was appropriately utilized. 2-Phenylimino-2-oxazolidine 3(Fig. 1),developed by Kim,has proven to be efficient in terms of stereoselectivity and yield in asymmetric alkylations [23]. Herein, we wish to report the preparation of a new camphor-based 2-phenylimino-2-oxazolidine chiral auxiliary and the preliminary results of our investigations concerning the asymmetric alkylations on this compound. The design of4(Fig. 1) was inspired by the results achieved with the 2-phenylimino-2-oxazolidine3and by the advantageous topological bias of the camphor skeleton to form conformationally rigid derivatives.

|
Download:
|
| Fig. 1. Examples of camphor-based auxiliaries and 2-phenylimino-2-oxazolidine auxiliaries. | |
All solvents were dried or purified by standard procedures before use. Separations by flash chromatography were performed on 300-400 mesh silica gel. Melting points were measured on a WRS-1A digital melting point apparatus. Optical rotations were measured using a sodium D line on a WZZ-2B Automatic Polarimeter. HPLC analyses were carried out on a Dionex chromatograph equipped with a diode-array UV detector. IR spectra were obtained on KBr pellets or CH2Cl2 solvent. NMR spectra were recorded on a WIPM-400 spectrometer in CD3COCD3 or CDCl3( 1H at 400 MHz and 13C at 100 MHz) using TMS as the internal standard. High-resolution mass spectra (HRMS) were recorded on an Agilent 1260-6224 LC-MS TOF using ESI (electrospray ionization).
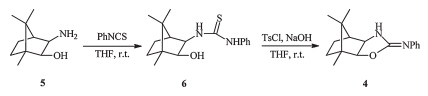
The synthesis of compound 6: To a solution of compound 5 (3.53 g,20.84 mmol) in THF (50 mL) was added phenyl isothiocyanate (2.96 mL,25.01 mmol) in THF (10 mL) dropwise,and the mixture was stirred at room temperature for 1 h. The solvent was removed under reduced pressure to afford the crude product, which was crystallized fromn-hexane/EtOAc (8:1,v/v) to give a white solid 6.
The synthesis of compound 4: To a solution of compound 6 (5.56 g,18.26 mmol) in THF (50 mL) were added NaOH (1.83 g, 45.70 mmol) andp-toluenesulfonyl chloride (3.83 g,20.09 mmol) in THF (50 mL) dropwise. Then the mixture was stirred at room temperature for 2 h,quenched with water (30 mL),and extracted with ether (50 mL×3). The organic layer was dried over MgSO4, filtered,and concentrated to afford the crude product,which was crystallized fromn-hexane/EtOAc (8:1,v/v) to give the yellow solid .
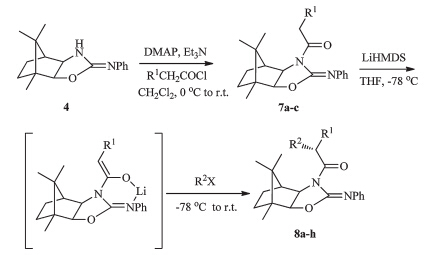
General procedure for the synthesis ofN-acylimides7a-c:Toa solution of chiral auxiliary 4(4.05 g,15.0 mmol) in CH2Cl2(50 mL) was added DMAP (0.37 g,3.0 mmol) and Et3N (2.58 mL, 17.81 mmol),and then the corresponding acyl chloride (19.40 mmol) in CH2Cl2 (10 mL) was added dropwise to the reaction mixture at 0℃. After stirring at 0℃ for 40 min,the reaction was quenched with saturated aqueous NH4Cl,and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (30 mL×3). The organic layers were combined,washed with saturated aqueous NaHCO3 and brine,dried over MgSO4, filtered,and concentrated. Purification of the crude product by silica gel chromatography (n-hexane/EtOAc,10:1,v/v) gave theNacylation products7a-c.
General procedure for asymmetric alkylations to afforded the alkylated products 8a-h: To a dry round-bottomed flask under nitrogen was added compound 7a-c(2.0 mmol) in anhydrous THF (20 mL). The solution was cooled to -78℃ and LiHMDS in THF (1.0 mol/L,2.0-3.0 equiv.) was added dropwise,and the solution was allowed to stir for 30 min. Then the mixture was treated with halides (3.0-5.0 equiv.). After stirring for 30 min at-78℃ and 1 h at 0℃,the reaction mixture was quenched with saturated aqueous NH4Cl (10 mL) and extracted with ether. The combined extracts were dried over MgSO4,filtered,and concentrated. HPLC analysis of the crude product revealed the isomer ratios. Purification by silica gel chromatography (n-hexane/EtOAc,10:1,v/v) afforded the alkylated products 8a-h.
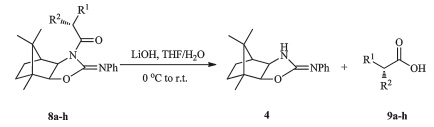
General procedure for the synthesis ofa-alkylated carboxylic acid 9a-h: The alkylated products 8a-h(1.0 mmol) were treated at 0℃ with LiOH (0.072 g,3.0 mmol) in a 1:4 mixture of THF/H2O (10 mL). The reaction was stirred overnight at room temperature and then concentrated under reduced pressure. The organic layer was extracted with CH2Cl2 (30 mL×3) and concentrated to recover quantitatively the chiral auxiliary 4. Acidification of the aqueous layer to pH 1,and extraction with EtOAc furnished the desireda-alkylated carboxylic acids 9a-h. 3. Results and discussion
As shown in Scheme 1,the camphor-based 2-phenylimino-2-oxazolidine chiral auxiliary 4 was readily prepared in two steps starting from exo-3-amino-exo-2-hydroxybornane 5,which was derived from natural (+)-camphor according to the literature procedure [24]. Exo-3-amino-exo-2-hydroxybornane 5 was treated with phenyl isothiocyanate to give the corresponding N-(2-hydroxyethyl) thiourea 6 in 93% yield,followed by cyclodesulfurization of the thiourea to give the camphor-based 2-phenylimino-2-oxazolidine 4 in 87% yield using p-toluenesulfonylchloride and NaOH.
To test the chemical and stereochemical behaviour of chiral auxiliary 4 for asymmetric synthesis,we evaluated the alkylation of N-acylimides 7a-c with representative alkyl halides. N-acylimides 7a-c were obtained in 85%-90% yields by treatment of chiral auxiliary 4 with acyl chloride in the presence of 4-(N,Ndimethylamino)pyridine (DMAP) and Et3N,and then the alkylations of N-acylimide 7a were first studied. Lithium enolate was formed by treating 7a with LiHMDS (2.0 equiv.) at-78℃ for 30 min and the subsequent addition of the alkyl halide (3.0- 5.0 equiv.) led to the formation of the correspondinga-alkylated products 8a-c(Scheme 2 and Table 1).

|
Download:
|
| Scheme 1.Synthesis of chiral auxiliaries. | |

|
Download:
|
| Scheme 2.TheN-acylation and the alkylation reactions. | |
| Table 1 Diastereoselective alkylations ofN-acylimides 7a-c. |
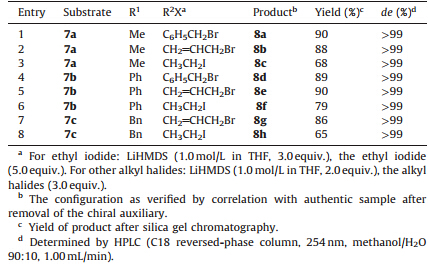
As shown in Table 1,all the alkylated products were obtained with excellent diastereomeric excess (de>99%). The alkylation reaction with benzyl bromide and allyl bromide using the Nacylimide 7a gave the required products 8a-b in 88%-90% yields (Table 1,entries 1 and 2),which are slightly higher than those obtained using the auxiliary3(82%-88%) [23]. The alkylation reaction with ethy l iodide afforded product 8cin higher yield (68%),as compared with the 55% obtained for the auxiliary3[23], but required 3.0 equiv. of base to complete the reaction (Table 1, entry 3).
We next investigated the alkylation reactions of theNacylimides 7b-c. The formation of the enolates was achieved with LiHMDS,followed by treatment with benzyl bromide and allyl bromide to give products 8d-e and 8g in high yields (86%-90%) and excellent diastereomeric excess (>99%) (Table 1,entries 4-5 and 7). Although a somewhat lower yield was observed with ethyl iodide,the diastereoselectivity was found to be virtually complete (Table 1,entries 6 and 8). All these results show that the steric hindrance of the camphor skeleton plays an important role in controlling the stereoselectivity,in a similar manner to that observed with the camphor derivatived auxiliaries 1 and 2.
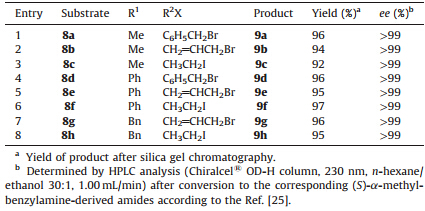
The elucidation of the configuration of thea-alkylated products8a-h was achieved by lithium hydroxide hydrolysis to afford the corresponding carboxylic acids 9a-h,along with the recovery of the chiral auxiliary4in nearly quantitative yield (Scheme 3 and Table 2). As expected,no products resulting from endocyclic cleavage were observed in the cleavage reaction. The absolute configurations of acids9a-hwere determined by comparing the measured optical rotations with the known values. The enatiomeric purities were determined by HPLC analysis of the corresponding (S)-a-methylbenzylamine-derived amides with Chiralcel ® OD-H column.

|
Download:
|
| Scheme 3.Hydrolysis of chiral auxiliaries. | |
| Table 2 Hydrolysis of the alkylated products 8a-h. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conclusion,we have developed a new chiral auxiliary, camphor-based 2-phenylimino-2-oxazolidine,which has great potential for asymmetric alkylations. The alkylated products were obtained in high yields with excellent diastereoselectivities. Moreover,the efficient workup after the hydrolysis reaction allowed a quantitative recovery of the chiral auxiliary and afforded the chirala-alkylated carboxylic acids in good yield and in almost enatiomerically pure form. Acknowledgments We gratefully acknowledge the National Natural Sciences Foundation of China (No. 21342002) and 2011 National Major Scientific Instrument and Equipment Development Project (No. 2011YQ12003505) for financial support. Appendix A. Supplementary data Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.05.044.
| [1] | J.C. Liu, Y.S. Yang, R.Y. Ji, A convenient method for the asymmetric synthesis of KAD-12293, Chin. Chem. Lett. 16 (2005) 430-432. |
| [2] | J.C. Nino, S. Mao, Y. Di Guan, Asymmetric synthesis of alpha-substituted-gammabutyrolactone empolying prolinol type of auxiliaries, Chin. Chem. Lett. 16 (2005) 17-19. |
| [3] | J.M. García, J.M. Odriozola, A. Lecumberri, J. Razkin, A. Gonzá lez, Concise and efficient route to the Alzheimer's therapeutic agent (R)-arundic acid, Tetrahedron 64 (2008) 10664-10669. |
| [4] | F. Colpaert, S. Mangelinckx, G. Verniest, N.D. Kimpe, Asymmetric synthesis of aalkylated N-sulfinyl imidates as new chiral building blocks, J. Org. Chem. 74 (2009) 3792-3797. |
| [5] | B.F. Li, R.M. Hughes, J. Le, et al., Efficient synthesis of (2S,3S)-2-ethyl-3-methylvaleramide using (1S,2S)-pseudoephedrine as a chiral auxiliary, Org. Process Res. Dev. 13 (2009) 463-467. |
| [6] | C.L. Hugelshofer, K.T. Mellem, A.G. Myers, Synthesis of quaternary a-methyl aamino acids by asymmetric alkylation of pseudoephenamine alaninamide pivaldimine, Org. Lett. 15 (2013) 3134-3137. |
| [7] | N. Satoh, S. Yokoshima, T. Fukuyama, Total synthesis of (-)-salinosporamide A, Org. Lett. 13 (2011) 3028-3031. |
| [8] | F. Fé court, G. Lopez, A.V.D. Lee, J. Martinez, G. Dewynter, Cyclosulfamide as a chiral auxiliary: application to efficient asymmetric synthesis (alkylation/aldolization), Tetrahedron: Asymmetry 21 (2010) 2361-2366. |
| [9] | G.B. Ren, Y.K. Wu, An aldol approach to the total synthesis of pamamycin 621 A, Org. Lett. 11 (2009) 5638-5641. |
| [10] | J.S. Zhang, C.F. Lu, Z.X. Chen, Y. Li, G.C. Yang, Boron enolates of a hydantoin chiral auxiliary derived from 1-phenylalanine: a versatile tool for asymmetric aldol reactions, Tetrahedron: Asymmetry 23 (2012) 72-75. |
| [11] | G.L. Khatik, R. Khurana, V. Kumar, V.A. Nair, Asymmetric induction by (S)-4-isopropyl- 1-phenylimidazolidin-2-thione in titanium-mediated aldol reactions and its application in enantioselective synthesis of (R)-baclofen, Synthesis (2011) 3123-3132. |
| [12] | Y.C. Luo, H.H. Zhang, Y. Wang, P.F. Xu, Synthesis of a-amino acids based on chiral tricycloiminolactone derived from natural (+)-camphor, Acc. Chem. Res. 43 (2010) 1317-1330. |
| [13] | P.F. Xu, S. Li, T.J. L, C.C. Wu, et al., Asymmetric synthesis of a,a-disubstituted aamino acids by diastereoselective alkylation of camphor-based tricyclic iminolactone, J. Org. Chem. 71 (2006) 4364-4373. |
| [14] | H.H. Zhang, X.H.Hu, X.Wu, Y.C. Luo, et al., A convenient route to enantiopure 3-aryl- 2 3-diaminopropanoic acids by diastereoselective Mannich reaction of camphorbased tricyclic iminolactone with imines, J. Org. Chem. 73 (2008) 3634-3637. |
| [15] | C. Palomo, M. Oiarbide, A. Gonz, J.M. Garcfa, et al., Exo,exo-2,3-diaminoborneolderived imidazolidinone as chiral auxiliary for asymmetric alkylations, Tetrahedron Lett. 37 (1996) 4565-4568. |
| [16] | C. Palomo, F. Berree, A. Lindent, J.M. Villalgordo, Exo,exo-2-amino-3-borneolderived oxazolidinone as a new chiral auxiliary for use in asymmetric transformations, J. Chem. Soc. Chem. Commun. (1994) 1861-1862. |
| [17] | C. Palomo, M. Oiarbide, A. Mielgo, et al., Alkylation of chiral a-hydroxy ketones derived from (1R)-(+)-camphor. An asymmetric variant of the classical acetylene route to carbonyl compounds, Org. Lett. 3 (2001) 3249-3252. |
| [18] | T.H. Yan, C.W. Tan, H.C. Lee, H.C. Lo, T.Y. Huang, Asymmetric aldol reactions: a novel model for switching between chelation- and non-chelation-controlled aldol reactions, J. Am. Chem. Soc. 115 (1993) 2613-2621. |
| [19] | K.H. Ahn, S. Lee, A. Lira, Asymmetric aldol reactions employing a camphor-derived chiral oxazinone auxiliary, J. Org. Chem. 57 (1992) 5065-5066. |
| [20] | M.P. Bonner, E.R. Thornton, 4-(Benzotriazol-1-yl)-6H-benzo[c]tetrazolo[1,5- e][1,2,5] triazepine. A new heterocyclic ring system formed by a novel benzotriazole ring opening, J. Am. Chem. Soc. 113 (1991) 1299-1301. |
| [21] | K.H. Ahn, A. Lim, S. Lee, Diastereoselective alkylation reactions employing a camphorbased chiral oxazinone auxiliary, Tetrahedron: Asymmetry 4 (1993) 2435-2436. |
| [22] | E.H. Krenske, K.N. Houk, D. Lim, et al., Origins of stereoselectivity in the aalkylation of chiral hydrazones, J. Org. Chem. 75 (2010) 8578-8584. |
| [23] | G.J. Lee, T.H. Kim, J.N. Kim, U. Lee, L-Valinol and L-phenylalaninol-derived 2- phenylamino-2-oxazolines as chiral auxiliaries in asymmetric alkylations, Tetrahedron: Asymmetry 13 (2002) 9-12. |
| [24] | K. Chen, S.J. Jeon, P.J. Walsh, W.A. Nugent, (2S)-( )-3-exo-(Morpholino)isoborneol[(-)-MIB] ([1R-(exo,exo)]-1,7,7-trimethyl-3-morpholin-4-yl-bicyclo[2,2,1] heptan-2-ol), Org. Synth. 82 (2005) 87-92. |
| [25] | T. Kotake, Y. Hayashi, S. Rajesh, et al., Design and synthesis of a new polymersupported evans-type oxazolidinone: an efficient chiral auxiliary in the solid-phase asymmetric alkylation reactions, Tetrahedron 61 (2005) 3819-3833. |