




Aziridines have been demonstrated to be valuable building blocks in organic synthesis due to their reactivity and versatility [1, 2, 3, 4, 5, 6, 7, 8, 9]. Their high ring strain energy endows them with high reactivity and enables easy ring cleavage with various nucleophiles. The use of halides as nucleophiles leads to the formation of b-haloamines,which are versatile synthetic intermediates for the synthesis of functional materials and biologically active compounds. The direct conversion of aziridines into the corresponding b-haloamines has previously been reported with a variety of halides. They are HCl [10, 11, 12, 13],ZnX2 (X = Cl,Br,I) [14],MgBr2 [15, 16],NaX (X = Br,I) [16],CeCl3.7H2O/NaI [17],indium trihalides [18],LiX/Amb15 (X = Cl,Br,I) [19],BF3.OEt2as a fluorine source [20],iodine/thiophenol [21],zirconyl chloride [22],PPh3/halogenating agent [23],tetrabutylammonium halides in the presence of b-cyclodextrin [24],an activated DMF complex [25] and BF3.OEt2/ tetraalkylammonium halides,respectively [26]. Although these limited synthetic approaches have made significant progress and provided some value in the past few years,most of these methodologies suffer from disadvantages such as narrow substrate scope,low regioselectivity,long reaction times,formation of other inseparable regioisomers,and high temperature. Hence,the study of efficient,highly regioselective and stereoselective ring openings of aziridines still remains important and challenging. At the same time,the discovery of novel catalysts and the development of new methods that make use of mild experimental conditions and readily available and inexpensive halides are highly desirable. Iron is one of the most abundant metals on earth,and consequently one of the most inexpensive and environmentally friendly ones to use. Its salts are more practical catalysts compared to traditional Lewis acids in several carbon-carbon bond forming reactions and have found wide applications in organic synthesis [27, 28]. Herein,we will report a highly regioselective and efficient Fe(III) halidepromoted ring-opening reactions ofN-tosylaziridines with readily available Fe(III) halides to giveb-haloamines in good to excellent yields with better substrate scope. 2. Experimental
Typical procedure for the synthesis of chloroamines: A reaction mixture of well-stirredN-tosylaziridine 1(0.2 mmol) and FeCl3 (40 mol% or 50 mol%) in CH2Cl2 (2.5 mL) was stirred at the specified temperature for a period of time under air atmosphere (Scheme 1). After the reaction was completed,which was monitored by TLC,the reaction mixture was concentrated under vacuum,and the resultant crude mixture was purified by column chromatography on silica gel with petroleum ether/ethyl acetate to give the corresponding pure chloroamines 2 and 3. The experimental procedure for the synthesis of bromoamines is the same as that for the synthesis of chloroamines.

|
Download:
|
| Scheme 1. Ring openings ofN-tosylaziridines 1 with FeCl3 | |
Typical procedure for the synthesis of 6a: A mixture of bbromoamine 4a(0.2 mmol) and NaNO2(0.3 mmol,1.5 equiv.) in DMSO (1.2 mL) was stirred under air atmosphere at 65°C for 4 h (Scheme 2). After the complete conversion monitored by TLC,the reaction mixture was diluted with water (10 mL) and extracted with ether (2×15 mL). The combined organic layers were dried over anhydrous Na2SO4,concentrated in vacuum and the resulting product was purified by column chromatography on silica gel (200-300 mesh,ethyl acetate/petroleum ether,1:4) to afford pure 6a.

|
Download:
|
| Scheme 2. Nucleophile substitution of 4a with NaNO2. | |
The physical and spectral data and spectra of 1H NMR,13C NMRand HRMS of all products are given in Supporting information. 3. Results and discussion
Initially,our aim was to identify the best halides through screening metal halides in terms of the ring openings of Ntosylaziridines [29],and we began our research by choosing Ntosylcyclohexylaziridine1aas the model substrate. Subsequently, a series of halides was screened under air atmosphere at room temperature,and the results are summarized in Table 1. When AlCl3 was used,the trace amount of product was detected (Table 1, entry 1). Compared with MgCl2 (75%),FeCl3 provided a better result (81%) (Table 1,entry 3). So,FeCl3 was selected as a better nucleophile that also served as an excellent catalyst for the ring openings.
| Table 1 Screening of reaction conditions for ring openings ofN-tosylcyclohexylaziridine 1a with anhydrous chloride source.a |
Then,a series of experimental parameters was examined. Different solvents such as CH2Cl2,THF and CH3CN were screened. No product was detected when this reaction was performed in THF or CH3CN (Table 1,entries 4,5). CH2Cl2was identified as the best solvent for this reaction (Table 1,entry 3). The investigation of the temperature indicated that room temperature was suitable (Table 1,entry 3 vs.6,7). Subsequently,the screening of the amount of FeCl3 was carried out. 81% yield was obtained when 2.0 equiv. of FeCl3 was adopted (Table 1,entry 3). Decreasing the amount of FeCl3 from 2.0 to 0.5 equiv. can dramatically increase the yield to 94% (Table 1,entry 10). Further improvement of the yield was not observed by using 0.4 equiv of FeCl3 (Table 1,entry 11). Besides,it was found that the reaction time can be reduced by increasing the solvent dosages from 2.0 to 2.5 mL (Table 1,entry 12 vs.11). At last,extensive screening showed the optimal reaction conditions were 0.2 mmol N-tosylcyclohexylaziridine 1a and 50 mol% FeCl3 in 2.5 mL CH2Cl2 under air atmosphere at room temperature for 1.5 h (Table 1,entry 14).
Having established the optimal conditions of this ring-opening reaction,we turned to a survey of the substrate scope and generality of this method. Similarly,FeCl3 and FeBr3reacted well to give the corresponding chloro and bromo amines,and this reaction worked equally well with both aliphatic and aromaticNtosylaziridines,and the corresponding products were provided in good to excellent yields with high regioselectivity (Table 2). The important feature of the reaction is its high regioselectivity. Both cyclic and acyclic alkenes-derived aliphatic N-tosylaziridines proceeded smoothly with FeCl3 and FeBr3 affording only one regioisomer 2or4,respectively,in excellent yields,and the formation of the other regioisomers 3or5was not observed (Table 2,entries 1-4). The ring opening reactions of cyclic Ntosylaziridines were completelyanti-stereoselective,giving only the trans isomers. Acyclic terminal N-tosylaziridines gave high regioselectivity with the formation of only one product,which demonstrates the predominant attack of the nucleophile at the less hindered terminal carbon [17, 18]. In terms of monosubstituted aromatic N-tosylaziridines,neither the electronic properties of the substituents on the aromatic ring nor the steric hindrance had obvious influence on the yields. Except for1n which gave two isomers with high regioselectivity (94/6) about FeBr3,otherN-tosylaziridines provided the single regioisomer3or 5by nucleophilic attack of the halide ion (FeCl3 and FeBr3)atthe benzylic position,and up to 99% yields were obtained within 0.5 h (Table 2,entries 5-12). It was possible that electronic factors predominated over the steric factors in this process. In addition, moderate yields and high regioselectivity could also be obtained with condensed-ringN-tosylaziridines (Table 2,entries 13-14). Unfortunately,it can be seen that disubstituted aromaticNtosylaziridines provided the two regioisomer products in good yields with bad regioselectivity due to the effect of spatial structure and the fact that the regioisomers could not be separated by column chromatography on silica gel (Table 2, entries 15,16).
| Table 2 Regioselective ring openings of variousN-tosylaziridines with anhydrous FeX3.a |

{kind=link}
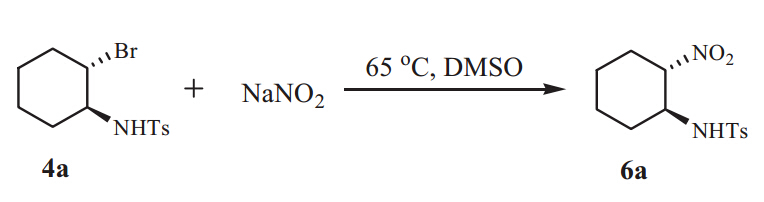{kind=link}
Br-substituted organic compounds have versatile applications in organic synthesis. Some new functional groups can be smoothly introduced by nucleophilic substitution of a bromo group. Furthermore,we examined the substitution reactions of thebhaloamines obtained through the ring openings ofN-tosylaziridines. The Br group in the product 4acould be conveniently substituted by the nitro group (-NO2) using NaNO2,and the desired product 6awas obtained in 55% yield (see Scheme 2). 4. Conclusion
In summary,we have developed a simple,practical,and highly efficient method for the synthesis ofb-haloamines through the ring-opening reactions of various N-tosylaziridines with readily available FeX3 (X = Cl,Br). More importantly,good to excellent yields and single regioisomers could be provided for a wide variety of mono-substituted substrates under extremely mild conditions. In addition,b-haloamines can be easily transferred into nitro compounds through the nucleophilic substitution of the Br group by NaNO2. Acknowledgments
We appreciate gratefully the Natural Science Foundation of Shanxi Province (Nos. 2012021007-2,2011011010-2) for financial support. The project is also supported by Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (No. 20120006). Appendix A. Supplementary data
Supplementary data associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.cclet.2014.03.033.
| [1] | P.F. Lu, Recent developments in regioselective ring opening of aziridines, Tetrahedron 66 (2010) 2549-2560. |
| [2] | G.S. Singh, M. D'hooghe, N. De Kimpe, Synthesis and reactivity of C-heteroatomsubstituted aziridines, Chem. Rev. 107 (2007) 2080-2135. |
| [3] | I.D.G. Watson, L. Yu, A.K. Yudin, Advances in nitrogen transfer reactions involving aziridines, Acc. Chem. Res. 39 (2006) 194-206. |
| [4] | X.E. Hu, Nucleophilic ring opening of aziridines, Tetrahedron 60 (2004) 2701-2743. |
| [5] | J.B. Sweeney, Aziridines: epoxides' ugly cousins? Chem. Soc. Rev. 31 (2002) 247-258. |
| [6] | W. McCoull, F.A. Davis, Recent synthetic applications of chiral aziridines, Synthesis (2000) 1347-1365. |
| [7] | P. Dauban, R.H. Dodd, 2,3-Aziridino-2,3-dideoxy-D-ribono-g-lactone 5-phosphonate: stereocontrolled synthesis from D-lyxose and unusual aziridine ring opening, J. Org. Chem. 62 (1997) 4277-4284. |
| [8] | G. Chouhan, H. Alper, Domino ring-opening/carboxamidation reactions of N-tosyl aziridines and 2-halophenols/pyridinol: efficient synthesis of 1,4-benzo-and pyrido-oxazepinones, Org. Lett. 12 (2010) 192-195. |
| [9] | G.D. Sala, A. Lattanzi, Highly enantioselective synthesis of β-amidophenylthioethers by organocatalytic desymmetrization of meso-aziridines, Org. Lett. 11 (2009) 3330-3333. |
| [10] | E. Leemans, S. Mangelinckx, N. De Kimpe, Novel synthesis of chiral unactivated 2-aryl-1-benzylaziridines, Synlett (2009) 1265-1268. |
| [11] | B. Crousse, S. Narizuka, D. Bonnet-Delpon, J.P. Bengue, First stereoselective synthesis of cis 3-CF3-aziridine-2-carboxylates. A route to new (trifluoromethyl) α-functionalised β-amino acids, Synlett (2001) 679-681. |
| [12] | C.A. Ray, E. Risberg, P. Somfai, Lewis acid-catalyzed hetero Diels-Alder cycloadditions of 3-alkyl, 3-phenyl and 3-carboxylated 2H-azirines, Tetrahedron Lett. 42 (2001) 9289-9291. |
| [13] | D. Gnecco, F.L. Orea, A. Galindo, et al., Regiospecific and enantiospecific ring opening of methyl (+)-(1'R, 2R)-and (-)-(1'R, 2S)-1-(2-phenylethanol) aziridine-2-carboxylates, Molecules 5 (2000) 998-1003. |
| [14] | M.K. Ghorai, K. Das, A. Kumar, K. Ghosh, An efficient route to regioselective opening of N-tosylaziridines with zinc(Ⅱ) halides, Tetrahedron Lett. 46 (2005) 4103-4106. |
| [15] | G. Righi, T. Franchini, C. Bonini, Highly regioselective opening of optically active N-Boc-2,3-aziridino alcohol derivatives with metal halides, Tetrahedron Lett. 39 (1998) 2385-2388. |
| [16] | C. Bonini, G. Righi, R. D'Achille, Regioselective opening of 3-substituted N-ethoxycarbonyl aziridine-2-carboxylates with metal halides toward the preparation of α and β-amino acids, Tetrahedron Lett. 37 (1996) 6893-6896. |
| [17] | G. Sabitha, R.S. Babu, M. Rajkumar, C.S. Reddy, J.S. Yadav, Highly regioselective ring opening of epoxides and aziridines using cerium(Ⅲ) chloride, Tetrahedron Lett. 42 (2001) 3955-3958. |
| [18] | J.S. Yadav, B.V.S. Reddy, G.M. Kumar, Indium trihalide mediated regioselective ring opening of aziridines: a facile synthesis of 2-haloamines, Synlett (2001) 1417-1418. |
| [19] | G. Righi, C. Potini, P. Bovicelli, Stereo-and regioselective ring opening of alkenyl aziridines with metal halides, Tetrahedron Lett. 43 (2002) 5867-5869. |
| [20] | C.H. Ding, L.X. Dai, X.L. Hou, An efficient and highly regioselective fluorination of aziridines using BF3·OEt2 as fluorine source, Synlett (2004) 2218-2220. |
| [21] | J. Wu, X.Y. Sun, W. Sun, S.Q. Ye, Unexpected highly efficient ring-opening of aziridines or epoxides with iodine promoted by thiophenol, Synlett (2006) 2489-2491. |
| [22] | B. Das, M. Krishnaiah, K. Venkateswarlu, Regio-and stereoselective ring opening of epoxides and aziridines using zirconyl chloride: an efficient approach for the synthesis of β-chlorohydrins and β-chloroamines, Chem. Lett. 36 (2007) 82-83. |
| [23] | M. Kumar, S.K. Pandey, S. Gandhi, V.K. Singh, PPh3/halogenating agent-mediated highly efficient ring opening of activated and non-activated aziridines, Tetrahedron Lett. 50 (2009) 363-365. |
| [24] | M. Narender, K. Surendra, N.S. Krishnaveni, M.S. Reddy, K.R. Rao, A facile regioselective ring opening of aziridines to haloamines using tetrabutylammoniumhalides in the presence ofb-cyclodextrin in water, Tetrahedron Lett. 45 (2004) 7995-7997. |
| [25] | M.K. Pandey, A. Bisai, V.K. Singh, Regioselective ring opening of aziridines with activated DMF complexes: a facile synthesis of β-haloamines, Tetrahedron Lett. 45 (2004) 9661-9663. |
| [26] | M.K. Ghorai, A. Kumar, D.P. Tiwari, BF3·OEt2-Mediated highly regioselective SN2-type ring-opening of N-activated aziridines and N-activated azetidines by tetraalkylammonium halides, J. Org. Chem. 75 (2010) 137-151. |
| [27] | Q.J. Li, M. Shi, C. Timmons, G.G. Li, FeCl3-Catalyzed aminohalogenation of arylmethylenecyclopropanes and arylvinylidenecyclopropanes and corresponding mechanistic studies, Org. Lett. 8 (2006) 625-628. |
| [28] | C. Bolm, J. Legros, J.L. Paih, L. Zani, Iron-Catalyzed reactions in organic synthesis, Chem. Rev. 104 (2004) 6217-6254. |
| [29] | (a) D. Kano, S. Minakata, M. Komatsu, Novel organic-solvent-free aziridination of olefins: chloramine-T-I2 system under phase-transfer catalysis conditions, J. Chem. Soc. Perkin Trans. 1 (2001) 3186-3188; (b) V.V. Thakur, A. Sudalai, N-Bromoamides as versatile catalysts for aziridination of olefins using chloramine-T, Tetrahedron Lett. 44 (2003) 989-992. |