




b College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai 201620, China
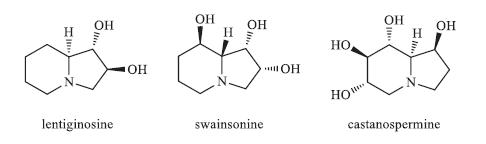
Polyhydroxylated indolizidine alkaloids are widely found in microorganisms,vertebrates,higher invertebrates,and plants [1]. Some of these aza fused bicyclic compounds,such as lentiginosine [2],swainsonine [3],and castanospermine [4],have been reported to exhibit good glycosidase inhibition activity,which makes them potential therapeutic agents (Fig. 1). Among them,castanospermine has attracted considerable attention. It is a strong inhibitor of a- and b-glucosidases [5] and has shown potential antitumor, antiviral,and immunomodulating activities [6]. Because of its unique biological activities,a lot of efforts have been devoted into the synthesis of castanospermine [7] as well as its analogues [8] to study their structure-activity relationship (SAR).

|
Download:
|
| Fig. 1.Polyhydroxylated indolizidines. | |
{kind=link}
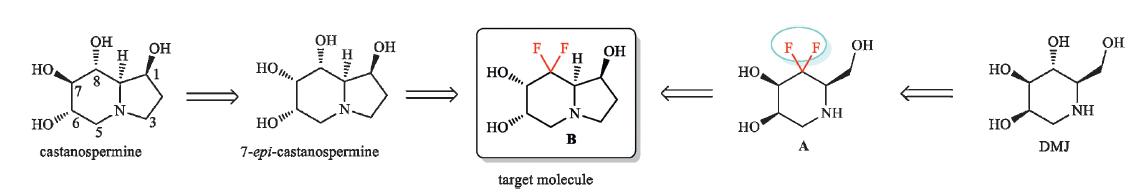
The major drawback for castanospermine in clinical applications is the low inhibition selectivity between a- and bglucosidases. In 1997,Tyler and co-workers reported that 7-epicastanospermine showed higher a/b glucosidase selectivity than castanospermine while retaining almost complete activity towards a-glucosidase (Scheme 1) [9]. To further improve the selectivity and the potency of the inhibition of glycosidases,it is highly desirable to develop new polyhydroxylated alkaloid analogues. In 2006,our group found that introduction of a gem-difluoromethylene group (CF2) into 1-deoxymannonojirimycin (DMJ) could improve the inhibition selectivity of compound A [10]. As a part of our interest in the synthesis of fluorinated iminosugars [10, 11],we designed a new gem-difluoromethylenated analogue B of 7-epicastanospermine. The strongly electron-withdrawing gem-difluoromethylene group would reduce the pKa value and might change the stereoconfiguration,thus affecting the inhibition activity and selectivity. In this paper,we present our results on the synthesis of this novel castanospermine analogue.

|
Download:
|
| Scheme 1.Design of target molecule B. | |
{kind=link}
To a stirring solution of (S)-O-benzylmalic acid dimethyl ester 2 (0.90 g,3.57 mmol) in CH2Cl2 (50 mL) was added magnesium bromide etherate (1.0 g,4.0 mmol) at 0 ℃. The solution was stirred for 1 h at 0 ℃ and cooled to -90 ℃. To the solution was added a 1.5 mol/L solution of diisobutylaluminum hydride in toluene (3.0 mL,4.5 mmol) via syringe pump over 90 min,then the reaction mixture was stirred at -90 ℃ for 2 h. Methanol (4 mL) was slowly added followed by saturated Rochelle salts (80 mL). The mixture was warmed to room temperature and stirred for 12 h. The layers were separated and the aqueous phase was extracted three times with methylene dichloride. The combined organic phase was dried with Na2SO4 and concentrated. The mixture was separated by flash column chromatography (petroleum ether/ethyl acetate = 8/1) to yield 0.61 g (77%) of a colourless oil. The colourless oil was dissolved in DMF (10 mL). After that, indium (474 mg,4.12 mmol) and 3-bromo-3,3-difluoropropane (420 mL,4.20 mmol) was added. The reaction mixture was stirred at room temperature for 24 h. The reaction mixture was then quenched with 1 mol/L HCl and extracted with CH2Cl2. The combined organic extract was washed with brine,dried over anhydrous Na2SO4,and filtered. The solvent was removed in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 8/1) to give 255 mg (31% yield) of compound 3a as a clear oil and 362 mg (44% yield) of compound 3b as a clear oil.
3a: [α]D20 -6:93 (c 1.00,CHCl3); 1H NMR (300 MHz,CDCl3): δ 7.31-7.30 (m,5H),6.08-5.95 (m,1H),5.76-5.68 (m,1H),5.54-5.47 (m,1H),4.67-4.60 (m,2H),4.28-4.25 (m,1H),3.76-3.64 (m,4H), 2.73-2.71 (m,2H),2.42 (br,1H); 19F NMR (282 MHz,CDCl3): δ -107.0 (dt,1F,∫ = 253.0 Hz,9.45 Hz),-111.5 (dt,1F,∫ = 252.4 Hz, 11.0 Hz); 13C NMR (100 MHz,CDCl3): δ 171.4,137.4,130.6 (t, ∫ = 25.7 Hz),128.5,128.1,120.7 (t,∫ = 9.5 Hz),119.4 (t, ∫ = 242.8 Hz),74.1 (t,∫ = 29.6 Hz),73.2,72.9,51.8,37.3; IR (thin film,cm-1): 3500,3033,2953,1738,1439; MS (ESI): m/z 301.3 (M+H+),318.3 (M+NH4 +); 323.2 (M+Na+); HRMS Calcd. for C15H18O4F2Na: 323.1065; Found: 323.1073.
3b: [α]D20 -15:17 (c 1.00,CHCl3); 1H NMR (300 MHz,CDCl3): d 7.40-7.30 (m,5H),6.04-5.87 (m,1H),5.80-5.74 (m,1H),5.63-5.60 (m,1H),4.65-4.51 (m,3H),4.46-4.44 (m,1H),2.87-2.78 (m,1H), 2.65-2.59 (m,1H); 19F NMR (282 MHz,CDCl3): δ -109.8 (ddd,1F, ∫ = 258.6 Hz,12.7 Hz,4.5 Hz),-113.0 (ddd,1F,∫ = 258.9 Hz, 18.6 Hz,10.6 Hz); 13C NMR (100 MHz,CDCl3): δ 174.2,136.7, 128.8 (t,∫ = 25.8 Hz),128.7,128.3,127.9,122.9 (t,∫ = 9.5 Hz),117.6 (t,∫ = 242.7 Hz),84.3 (t,∫ = 31.4 Hz),73.4,71.4,34.9; IR (thin film, cm-1): 3033,2932,1691,1455,1208; MS (ESI): m/z 286.2 (M+NH4 +); HRMS Calcd. for C14H14O3F2Na: 291.0803; Found: 291.0811. 2.2. (3S,4S)-3-(Benzyloxy)-5,5-difluorohept-6-ene-1,4-diol (4)
To a suspension of LiAlH4 (91.2 mg,2.4 mmol) in dry THF (5 mL) was added a solution of 3b (0.36 g,1.2 mmol) in dry THF at 0 ℃. The mixture was stirred for 3 h at 0 ℃ and quenched by careful addition of water and then extracted with CH2Cl2. The combined organic layer was washed with brine,dried over anhydrous Na2SO4,and concentrated in vacuo. The residue was quickly purified by silica gel column chromatography (petroleum ether/ ethyl acetate = 2/1) to afford compound 4 (0.28 g,86%) as a clear oil. [α]D20 -17:01 (c 1.00,CHCl3); 1H NMR (300 MHz,CDCl3): δ 7.35- 7.25 (m,5H),6.10-5.93 (m,1H),5.73-5.68 (m,1H),5.50-5.47 (m, 1H),4.52 (dd,2H,∫ = 17.4 Hz,11.4 Hz),4.07-3.98 (m,1H),3.90- 3.63 (m,3H),2.96 (br,2H),2.02-1.92 (m,2H); 19F NMR (282 MHz, CDCl3): δ -107.7 (dt,1F,∫ = 251.5 Hz,10.3 Hz),-109.3 (dt,1F, ∫ = 251.8 Hz,12.0 Hz); 13CNMR (100 MHz,CDCl3): δ 137.6,130.9 (t, ∫ = 25.4 Hz),128.6,128.1,128.0,120.4 (t,∫ = 9.5 Hz),119.7 (t, ∫ = 243.1 Hz),76.3,73.8 (t,∫ = 27.9 Hz),71.8,58.9,31.7; IR (thin film,cm-1): 3392,3033,2928,1664,1056; MS (ESI): m/z 295.2 (M+Na+); HRMS Calcd. for C14H18O3F2Na: 295.1116; Found: 295.1120. 2.3. (2R,3S)-1-Allyl-3-(benzyloxy)-2-(1,1-difluoroallyl)pyrrolidine (5)
To a solution of compound 4 (1.0 g,3.68 mmol) in anhydrous CH2Cl2 (12 mL),and NEt3 (3.4 mL,23.8 mmol),MsCl (1.3 mL, 16.7 mmol) was added slowly at 0 ℃. The reaction mixture was then warmed to room temperature and stirred overnight. The reaction was quenched with water. The resulting mixture was extracted with CH2Cl2. The combined organic layer was washed with brine,dried over anhydrous Na2SO4,and filtered. The solvent was removed in vacuo. The residue was dissolved in allylamine (6 mL). Then the reaction mixture was heated to 145 ℃ in the sealeδ tub for 10 h. The allylamine was removed in vacuo and then the residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 15/1) to give 870 mg (81%,yield, two steps) of compound 5 as a clear oil. [α]D20 -16:86 (c 1.10, CHCl3); 1H NMR (300 MHz,CDCl3): δ 7.34-7.25 (m,5H),6.35-6.17 (m,1H),5.93-5.79 (m,1H),5.68-5.62 (m,1H),5.42-5.38 (m,1H), 5.19-5.09 (m,2H),4.53 (s,2H),4.12 (q,1H,∫ = 6.3 Hz),3.57-3.51 (m,1H),3.18-3.00 (m,3H),3.17 (q,1H,∫ = 8.7 Hz),1.99-1.92 (m, 2H); 19FNMR(282 MHz,CDCl3): d-96.4 (d,1F,∫ = 255.2 Hz),-97.3 (dt,1F,∫ = 254.9 Hz,13.25 Hz); 13C NMR (100 MHz,CDCl3): δ 138.2, 134.8,133.0 (t,∫ = 24.5 Hz),128.3,127.7,127.6,121.6 (t, ∫ = 239.9 Hz),119.3 (t,∫ = 9.65 Hz),117.4,79.1 (d,∫ = 7.0 Hz), 72.1,68.7 (dd,∫ = 30.3 Hz,25.6 Hz),58.2,50.0 (d,∫ = 6 Hz),30.4; IR (thin film,cm-1): 3066,2924,2359,1419,1354,1121,738; MS (ESI): m/z 298.4 (M+H+); HRMS Calcd. for C17H22OF2N: 294.1664; Found: 294.1657. 2.4. (1S,8aR)-1-(Benzyloxy)-8,8-difluoro-1,2,3,5,8,8ahexahydroindolizine (6)
To a solution of compound 5 (520 mg,1.77 mmol) in anhydrous toluene (50 mL) was added Grubbs’ II catalyst (70 mg, 0.0824 mmol). The reaction mixture was then warmed to 80 ℃ and stirred for 10 h. After the solvent was evaporated,the crude product was purified by flash silica gel column chromatography (petroleum ether/ethyl acetate = 6/1) to give compound 6 (375 mg, 80%) as a clear oil. [α]D20 -98:66 (c 1.15,CHCl3); 1H NMR (300 MHz,CDCl3): δ 7.40-7.23 (m,5H),6.15-6.11 (m,1H),5.92- 5.86 (m,1H),4.66 (dd,2H,∫ = 37.5 Hz,12.3 Hz),4.39-4.33 (m,1H), 3.61-3.55 (m,1H),3.28 (t,1H,∫ = 8.6 Hz),3.00-2.93 (m,1H),2.73- 2.64 (m,1H),2.36 (q,1H,∫ = 8.4 Hz),2.26-2.15 (m,1H),2.07-1.95 (m,1H); 19F NMR (282 MHz,CDCl3): δ -98.3 (d,1F,∫ = 270.4 Hz), -100.5 (d,1F,∫ = 269.3 Hz); 13C NMR (100 MHz,CDCl3): δ 138.7, 133.8 (t,∫ = 9.8 Hz),128.3,127.4,127.3,123.7 (dd,∫ = 30.3 Hz, 26.4 Hz),117.7 (dd,∫ = 244.8 Hz,229.7 Hz),78.2,72.2,65.9 (d, ∫ = 20.7 Hz),65.6 (d,∫ = 21.3 Hz),51.6,31.2; IR (thin film,cm-1): 3033,2987,1620,1237,987; MS (ESI): m/z 266.3 (M+H+); HRMS Calcd. for C15H17OF2NNa: 288.1170; Found: 288.1164. 2.5. (3S,4S)-3-(Benzyloxy)-5,5-difluoro-4-hydroxyhept-6-enyl acetate (7)
To a solution of compound 4 (1.032 g,3.79 mmol) in vinyl acetate (15 mL) was added lipase AK (0.52 g). The solution was stirred at room temperature. After 24 h,the solution was filtered and volatiles were removed by reduced pressure. The crude product was chromatographed (petroleum ether/ethyl acetate = 8/1) to afford 7 (1.037 g,87%) as a clear oil. [α]D20 -35:71 (c 1.00,CHCl3); 1H NMR (400 MHz,CDCl3): δ 7.35-7.25 (m,5H), 6.05-5.92 (m,1H),5.69 (d,1H,∫ = 17.2 Hz),5.49 (d,1H, ∫=11.2Hz),4.51 (dd,2H,∫ = 34.4 Hz,11.2 Hz),4.23-4.12 (m, 2H),4.03 (td,1H,∫ = 11.2 Hz,3.2 Hz),3.77-3.74 (m,1H),2.04-1.91 (m,5H); 19F NMR (282 MHz,CDCl3): δ -107.5 (dt,1F,∫ = 251.5 Hz, 11.3 Hz),-109.4 (dt,1F,∫ = 251.3 Hz,11.3 Hz); 13C NMR (100 MHz,CDCl3): δ 171.1,137.4,130.5 (t,∫ = 25.3 Hz),128.5, 128.1,128.0,120.6 (t,∫ = 9.7 Hz),119.4 (dd,∫ = 241.2 Hz, 243.3 Hz),74.6 (t,∫ = 1.5 Hz),73.6 (t,∫ = 28.3 Hz),72.0,61.1, 28.7,20.9; IR (thin film,cm-1): 3466,3032,2904,1736,819; MS (ESI): m/z 315.0 (M+H+); HRMS Calcd. for C16H20O4F2NNa: 337.1222; Found: 337.1223. 2.6. (3S,4R)-4-Azido-3-(benzyloxy)-5,5-difluorohept-6-enyl acetate (8)
compound 7 (2.62 g,8.34 mmol) was dissolved in dry CH2Cl2 (50 mL). After that,DMAP (2.03 g,16.68 mmol) was added. The resulting mixture was cooled to -35 ℃. Then,Tf2O (2.10 mL, 12.52 mmol) was added dropwise to the solution with stirring. After that,the reaction mixture was stirred for about 3 h at 0 ℃. Water and NaHCO3 solution were added successively after the mixture was warmed to room temperature. Then the mixture was extracted with CH2Cl2,dried over anhydrous Na2SO4. The mixture was separated by flash column (petroleum ether/ethyl acetate = 8/1) to yield a colourless oil. The colourless oil was dissolved in DMF (20 mL). Then,sodium azide (2.7 g,41.7 mmol) was added carefully with stirring at 0 ℃ in an ice bath. The reaction mixture was stirred overnight at room temperature. Water was added to quench the reaction. The aqueous phase was extracted with CH2Cl2. The combined organic layer was washed with brine,dried over anhydrous Na2SO4,and concentrated in vacuo. The residue was quickly purified by silica gel column chromatography (petroleum ether/ethyl acetate = 8/1) to afford compound 8 (1.95 g,69% yield) as a clear oil. [α]D20 -45:97 (c 1.00,CHCl3); 1H NMR (300 MHz,CDCl3): δ 7.35-7.25 (m,5H), 6.08-5.95 (m,1H),5.78 (dt,1H,∫ = 17.4 Hz,2.1 Hz),5.49 (d,1H, ∫ = 11.1 Hz),4.59 (dd,2H,∫ = 39.3 Hz,11.1 Hz),4.24-4.08 (m, 2H),3.92-3.86 (m,1H),3.58-3.49 (m,1H),2.03-1.97 (m,5H); 19F NMR (282 MHz,CDCl3): δ -97.98 (d,1F,∫ = 253.2 Hz), -104.70 (d,1F,∫ = 251.3 Hz); 13C NMR (100 MHz,CDCl3): d 170.3,136.9,129.8 (t,∫ = 25.1 Hz),128.0,127.6,127.5,120.7 (t, ∫ = 9.6 Hz),119.0 (dd,∫ = 244.7 Hz,243.6 Hz),73.8 (d,∫ = 2.7 Hz), 72.8,66.7 (t,∫ = 28.3 Hz),60.1,31.0,20.4; IR (thin film,cm-1): 3032,2962,2114,1740,1240,989; MS (ESI): m/z 357.1 (M+NH4 +); HRMS Calcd. for C16H19O3F2N3Na: 362.1287; Found: 362.1287. 2.7. (3S,4R)-4-Azido-3-(benzyloxy)-5,5-difluorohept-6-en-1-ol (9)
compound 8 (0.87 g,4.06 mmol) was dissolved in MeOH (15 mL). After that,KOH (0.12 g) was added. The resulting mixture was stirred for about 1 h. Water was added and the aqueous phase was extracted with CH2Cl2. Then,the combined organic layers were washed with brine. After the resultant solution was dried over anhydrous Na2SO4 and filtered,the solvent was removed in vacuo. The residue was purified by flash silica gel column chromatography (petroleum ether/ethyl acetate = 6/1) to give 9 (713 mg,96%) as a clear oil. [α]D20 -49:66 (c 1.00,CHCl3); 1H NMR (400 MHz,CDCl3): δ 7.37-7.27 (m,5H), 6.09-5.96 (m,1H),5.77 (d,1H,∫ = 17.2 Hz),5.54 (d,1H, ∫ = 11.1 Hz),4.62 (dd,2H,∫ = 36.8 Hz,26.0 Hz),4.02-3.98 (m, 1H),3.73 (t,2H,∫ = 5.6 Hz),3.62-3.56 (m,1H),1.92 (q,2H, ∫ = 6.0 Hz),1.73 (br,1H); 19F NMR (282 MHz,CDCl3): δ -98.6 (dt, 1F,∫ = 253.5 Hz,9.9 Hz),-105.24 (dt,1F,∫ = 251.5 Hz,11.8 Hz); 13C NMR (100 MHz,CDCl3): δ 137.5,130.3 (t,∫ = 25.3 Hz),128.5, 128.0,127.9,121.1 (t,∫ = 9.7 Hz),119.5 (t,∫ = 244.2 Hz),75.0 (d, ∫ = 3.7 Hz),73.2,67.3 (t,∫ = 28.3 Hz),59.1,34.8 (d,∫ = 1.5 Hz); IR (thin film,cm-1): 3387,2929,2883,2113,989; MS (ESI): m/z 268.0 ([M-N2-H]+); HRMS Calcd. for C14H16O2F2N: 268.1149; Found: 268.1152. 2.8. 1-((2R,3S)-3-(Benzyloxy)-2-(1,1-difluoroallyl)pyrrolidin-1- yl)prop-2-en-1-one (10)
A solution of compound 9 (3.897 g,13.12 mmol) in dry CH2Cl2 (30 mL) was cooled to 0 ℃. Et3N (3.975 g,39.36 mmol),DMAP (80 mg,0.656 mmol),and MsCl (5 mL,65.6 mmol) were added. The mixture was stirred at room temperature for 12 h and then quenched with water. The two layers were separated and the aqueous layer was extracted with CH2Cl2. The combined organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 6/1) to give methanesulfonate (4.543 g, 12.08 mmol) as a clear oil. To a solution of methanesulfonate in THF (50 mL) was added Ph3P (4.75 g,18.12 mmol) and water (4 mL). The reaction mixture was warmed to 80 ℃ and stirred for 4 h and then the reaction mixture was monitoreδ by TLC. When the starting material was consumed,10% NaOH (aq.,15 mL) was added and the reaction mixture was stirred for 12 h at room temperature. The reaction mixture extracted with ethyl acetate. The combined organic layer was washed with water and dried over Na2SO4. The residue was dissolved in CH2Cl2 (20 mL). Then,K2CO3 (3.33 g, 24.16 mmol) and acryloyl chloride (2.18 g,24.16 mmol) was added. The mixture was stirred at room temperature for 12 h and then quenched with water. The two layers were separated,and the aqueous layer was extracted with CH2Cl2. The combined organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 10/1) to give compound 10 (2.54 g,63%) as a clear oil. [α]D20 -56:83 (c 2.00,CHCl3); 1H NMR (400 MHz,CDCl3): δ 7.34-7.26 (m,5H),6.57-6.51 (m,0.5H),6.41-6.37 (m,1.5H), 6.23-6.07 (m,1H),5.74-5.66 (m,2H),5.44 (dd,1H,∫ = 21.2, 11.2 Hz),4.95-4.87 (m,0.5H),4.70-4.63 (m,1H),4.55-4.49 (m,1H), 4.45-4.38 (m,0.5H),4.25-4.08 (m,1H),3.74-3.64 (m,1H),3.57- 3.46(m,1H),2.37-2.12 (m,2H); 19FNMR (376 MHz,CDCl3): d-96.2 (dd,0.48F,∫ = 252.3 Hz,10.9 Hz),-102.7 (dt,0.52F,∫ = 249.3 Hz, 13.9 Hz),-102.8 (dt,0.48F,∫ = 250.8 Hz,10.9 Hz),-103.4 (dt,0.52F, ∫ = 249.3 Hz,12.4 Hz); 13C NMR (100 MHz,CDCl3): δ 166.1,137.5, 137.3,132.4 (t,∫ = 24.6 Hz),131.9 (t,∫ = 24.6 Hz),129.1,128.5,128.4, 128.2,127.9,127.8,127.7,127.6,127.5,120.7 (t,∫ = 11.1 Hz),119.5 (t,∫ = 9.6 Hz),118.3 (t,∫ = 245.3 Hz),117.4 (t,∫ = 244.9 Hz),77.8,76.5, 72.7,61.4 (t,∫ = 23.1 Hz),58.1 (t,∫ = 26.8 Hz),44.0,43.1,29.5,27.5; IR (thin film,cm-1): 3030,2896,1655,1614,1421; MS (ESI): m/z 308 (M+H+); HRMS Calcd. for C17H19O2F2NNa: 330.1276; Found: 330.1270. 2.9. (1S,8aR)-1-(Benzyloxy)-8,8-difluoro-2,3,8,8a-tetrahydroindolizin- 5(1H)-one (11)
compound 10 (643 mg,0.31 mmol) and titanium isopropoxide (180 mg,0.643 mmol) in dry toluene (20 mL) was refluxeδ for 3 h under an argon atmosphere. Then Grubbs’ II catalyst dissolved in toluene (5 mL) was added dropwise to the mixture. The reaction mixture was stirred at reflux for 10 h. The reaction mixture was cooleδ to room temperature and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate = 8/1) to give compound 11 (450 mg,78% yield) as a yellow oil. [α]D20 -716:00 (c 0.51, CHCl3); 1H NMR (300 MHz,CDCl3): δ 7.33-7.24 (m,5H),6.52-6.47 (m,1H),6.20 (d,1H,∫ = 7.8 Hz),4.63 (dd,2H,∫ = 19.5 Hz,9.0 Hz), 4.48 (d,1H,∫ = 1.8 Hz),3.97-3.88 (m,1H),3.77-3.65 (m,2H),2.21- 2.15 (m,1H),1.93-1.84 (m,1H); 19F NMR (376 MHz,CDCl3): d -101.4 (dd,1F,∫ = 254.9 Hz,25.6 Hz),-106.4 (dt,1F,∫ = 273.7 Hz, 8.3 Hz); 13C NMR (100 MHz,CDCl3): δ 161.0 (d,∫ = 3.3 Hz),131.7, 133.7 (dd,∫ = 32.6 Hz,24.3 Hz),131.0 (dd,∫ = 11.3 Hz,8.3 Hz), 128.4,127.8,127.5,116.2 (dd,∫ = 251.3 Hz,231.5 Hz),77.85,72.2 (d,∫ = 1.5 Hz),62.8 (dd,∫ = 35.7 Hz,24.3 Hz),43.0,29.9; IR (thin film,cm-1): 2952,1675,1616,1445,1206; MS (ESI): m/z 302.0 (M+Na+),280.0 (M+H+); HRMS Calcd. for C15H16O2F2N: 280.1144; Found: 280.1154. 2.10. (1S,6R,7S,8aR)-1-(Benzyloxy)-8,8-difluoro-6,7-dihydroxyhexahydroindolizin- 5(1H)-one (12)
To a solution of compound 11 (450 mg,1.61 mmol) in acetone (5 mL) was added NMNO (438 mg,3.23 mmol),followed by addition of water (10 mL) at room temperature with stirring. Then a catalytic amount of OsO4 (5 mol %) solution in water (4% solution) was added. After the reaction mixture was stirred at room temperature for 48 h,it was quenched with saturated NaHSO3 solution and extracted with ethyl acetate. The combined organic layer was washed with brine,dried over anhydrous Na2SO4,and filtered. The solvent was removed in vacuo. The residue was purified by silica gel column chromatography (petroleum ether/ ethyl acetate = 2/1) to give 402 mg (80% yield) of compound 12 as a yellow oil. [α]D20 -93:56 (c 1.00,CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.37-7.27 (m,5H),4.63 (dd,2H,∫ = 14.7 Hz,9.0 Hz), 4.40-4.32 (m,2H),4.32 (s,1H),4.15 (d,1H,∫ = 19.5 Hz),3.69-3.65 (m,2H),2.21-2.15 (m,1H),1.97-1.92 (m,1H); 19F NMR (376 MHz, CDCl3): δ -113.6 (dd,1F,∫ = 256.0 Hz,8.3 Hz),-117.7 (dd,1F, ∫ = 256.0 Hz,25.6 Hz); 13C NMR (100 MHz,CDCl3): δ 169.0,137.7, 128.4,127.8,127.5,118.6 (t,∫ = 248.6 Hz),76.9,71.9 (d,∫ = 1.5 Hz), 70.3 (dd,∫ = 32.8,21.6 Hz),69.1 (d,∫ = 8.2 Hz),59.9 (dd,∫ = 37.2, 19.4 Hz),43.4,29.6; IR (thin film,cm-1): 3372,2896,1644,1117, 1068; MS (ESI): m/z 314.0 (M+H+); HRMS Calcd. for C15H18O4F2N: 314.1198; Found: 314.1212. 2.11. (1S,6S,7S,8aR)-1-(Benzyloxy)-8,8-difluorooctahydroindolizine- 6,7-diol (13)
To a stirred,cooled (0 ℃,ice bath) solution of 12 (185 mg, 0.585 mmol) in THF (3 mL) was added borane dimethylsulfide complex (2.0 mol/L in THF,3.0 mL,6.0 mmol). After 30 min,the cold bath was removed,and the reaction was heated to reflux and then stirred for 20 h. The reaction was then quenched with MeOH (1 mL) and concentrated under reduced pressure. The residue was dissolved in MeOH (5 mL). Then,the reaction was heated to reflux and stirred for 12 h. The mixture was concentrated under reduced pressure and the residue purified by column chromatography on silica gel (MeOH/CH2Cl2 = 1/50) to afford 13 (147 mg,85%) as a white solid. [α]D20 -18:06 (c 1.00,CD3OD); 1H NMR (400 MHz, CD3OD): δ 7.34-7.20 (m,5H),4.85 (s,2H),4.53 (dd,2H,∫ = 11.1 Hz, 9.3 Hz),4.28-4.25 (m,1H),3.93-3.84 (m,2H),3.14-3.10 (m,1H), 2.93-2.90 (m,1H),2.66 (dd,1H,∫ = 19.2 Hz,3.3 Hz),2.33 (t,1H, ∫ = 8.4 Hz),2.26-2.15 (m,2H),1.93-1.89 (m,1H); 19F NMR (376 MHz,CD3OD): δ -114.5 (d,1F,∫ = 253.5 Hz),-117.1 (dd, 1F,∫ = 254.5 Hz,26.0 Hz); 13C NMR (100 MHz,CD3OD): δ 138.5, 127.6,127.2,127.0,120.0 (t,∫ = 243.8 Hz),76.9,71.3,70.9 (dd, ∫ = 32.6 Hz,20.5 Hz),66.6 (d,∫ = 6.8 Hz),63.2 (dd,∫ = 30.4 Hz, 19.0 Hz),51.9,51.3,30.8; IR (thin film,cm-1): 3531,2952,1116, 1049,740; MS (ESI): m/z 300.0 (M+H+); HRMS Calcd. for C15H19O3F2NNa: 322.1225; Found: 322.1212. 2.12. 7-epi-8,8-Difluorocastanospermine (B)
HCOOH (1.045 mL) was added to a mixture of compound 13 (25 mg,0.084 mmol) and 10% Pd/C (334 mg) in MeOH (5 mL) under an argon atmosphere. The suspension was stirred for 4 h and then filtered through a short pad of celite. The filtrate was concentrated under reduced pressure and the residue was dissolved in water and passed through a column of ion-exchange resin (Dowex 1 × 8,OH-form) eluting with MeOH. The eluent was concentrated under reduced pressure to give 7-epi-8,8-difluorocastanospermine (B) (16 mg,92%) as a colourless solid. [α]D20 -20:52 (c 0.50,CD3OD); 1H NMR (400 MHz,CD3OD): d 4.47 (s,1H),3.91-3.89 (m,1H),3.82 (s,1H),3.13 (t,1H,∫ = 3.3 Hz), 2.93-2.91 (m,1H),2.51 (d,1H,∫ = 12.0 Hz),2.33-2.22 (m,2H),2.15 (q,1H,∫ = 8.8 Hz),1.77-1.69 (m,1H); 19F NMR (376 MHz,CD3OD): d-112.6 (d,1F,∫ = 251.9 Hz),-113.8 (dd,1F,∫ = 250.3 Hz,22.9 Hz); 13C NMR (100 MHz,CD3OD): δ 120.2 (t,∫ = 247.5 Hz),70.8 (dd, ∫ = 32.6 Hz,21.3 Hz),69.3,66.9 (d,∫ = 6.8 Hz),63.8 (dd,∫ = 28.8 Hz, 19.0 Hz),51.9,51.1,33.1; IR (thin film,cm-1): 3401,3321,2938, 1079,1057; MS (ESI): m/z 209.9 (M+H+); HRMS Calcd. for C8H14O3F2N: 210.0936; Found: 210.0946. 3. Results and discussion
The retrosynthetic analysis of target molecule B is shown in Scheme 2. compound B could be prepared by substrate-controlled cis-dihydroxylation of cycloalkene C. The six-member ring in compound C could be constructed by ring-closing metathesis (RCM) reaction from diene D,in which the pyrrolidine ring is easily accessible by intermolecular cyclization of allylamine and compound E. compound E is expected to be obtained by coupling of 3-bromo-3,3-difluoropropene and aldehyde F.

|
Download:
|
| Scheme 2.Retrosynthestic analysis of target molecule B. | |
{kind=link}
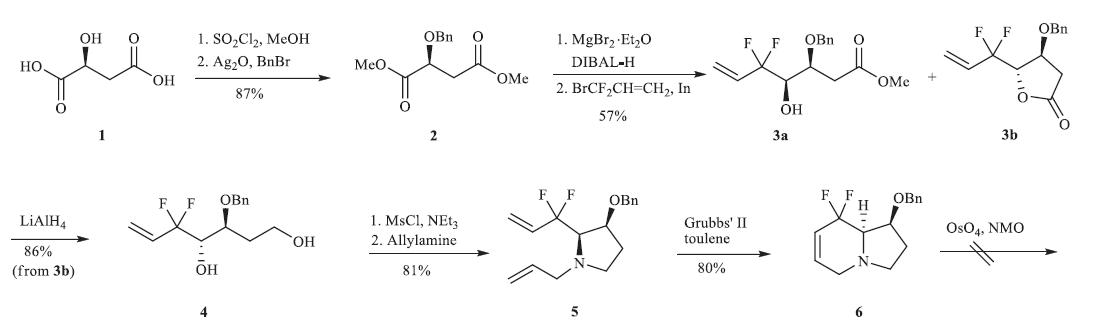
Our initial route towards compound B commenced from the cheap and commercially available L-(-)-malic acid 1 (Scheme 3). Esterfication of compound 1 with SO2Cl2/MeOH and then protection of the hydroxyl group under the condition of Ag2O/ BnBr gave compound 2 in high yield. Lewis acid mediated selective reduction of compound 2 produced the desired aldehyde [12], which reacted with 3-bromo-3,3-difluoropropene in the presence of indium affording two diastereoisomers 3a and 3b. Interestingly, compound 3b was obtained in the form of lactone. It was converted into diol 4 in high yield by reduction with LiAlH4. Reaction of both of the hydroxyl groups in diol 4 with MsCl and subsequent intramolecular cyclization with allylamine affordeδ the RCM reaction precursor 5. The RCM reaction of compound 5 with Grubbs’ II catalyst proceeded smoothly to afford alkene 6 in 80% yield. To our disappointment,the dihydroxylation of alkene 6 could not be achieved,despite trying many different reaction systems [13]. The failure of the dihydroxylation reaction might be ascribed to the coordination of nitrogen atom to the catalyst OsO4 [14].

|
Download:
|
| Scheme 3.Initial synthetic route. | |
{kind=link}
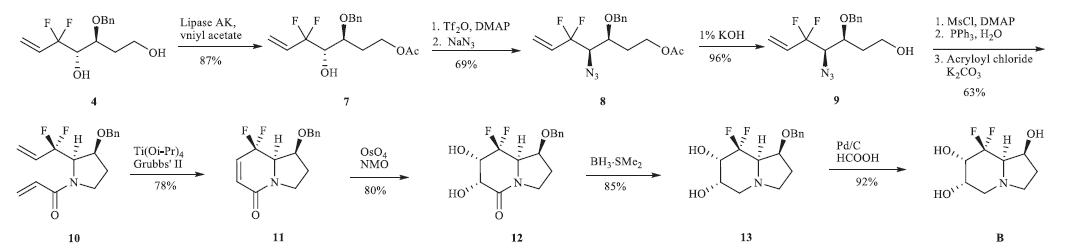
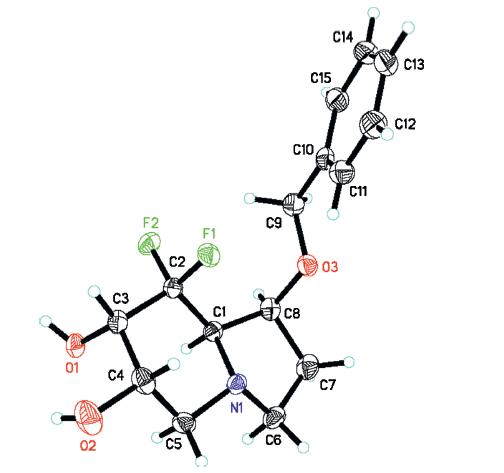
To reduce the coordination ability of nitrogen to osmium,we decided to connect an electron-withdrawing group to the nitrogen atom. The modified synthetic route is shown in Scheme 4. Selective protection of the primary hydroxyl group in compound 4 with vinyl acetate and Pseudomonas (AK) [15] gave the secondary alcohol 7 in 87% yield. Reaction of alcohol 7 with Tf2O in presence of DMAP afforded the corresponding triflate,which then reacted with NaN3 to give azide 8. Cleavage of the O-Ac group in a methanolic solution of 1% KOH afforded the desired alcohol 9. Mesylation of the hydroxyl group in compound 9 gave the corresponding methanesulfonate. Reduction of azide group with triphenylphosphine and subsequent intramolecular substitution cyclization afforded the pyrrolidine intermediate,which then was directly treated with acryloyl chloride to give diene 10 in 63% overall yield. Considering the high electron-deficient properties of diene 10,we performed the ring-closing metathesis (RCM) reaction under the reaction conditions developed by our group [16],with Grubbs’ II catalyst and co-catalyst Ti(i-PrO)4,affording the desired a,b-unsaturated lactam 11 in 78% yield. Dihydroxylation of lactam 11 catalyzed by OsO4 proceeded well giving diol 12 as a single isomer. The high diastereoselectivity can be explained by the steric hindrance of benzyl ether. Subsequent reduction of lactam 12 with borane dimethyl sulfide complex gave indolizidine 13 in 85% yield. The absolute configuration of 13 was confirmed by single-crystal X-ray diffraction analysis (Fig. 2). Finally,removal of the benzyl group,via hydrogenolysis in the presence of 10% Pd/C,provided the target molecule 7-epi-8,8-difluorocastanospermine B.

|
Download:
|
| Scheme 4.Modified synthetic route. | |
{kind=link}

|
Download:
|
| Fig. 2.X-ray crystallographic structures of compound 13. | |
{kind=link}
The synthesized 7-epi-8,8-difluorocastanospermine B was evaluated for its inhibitory activities against a-glucosidase from baker’s yeast and b-glucosidase from almonds. Unfortunately no significant inhibitory activity was observed. 4. Conclusion In conclusion,we have designed and prepared a novel gemdifluoromethylenated castanospermine analogue B. Intramolecular cyclization reaction was applied to construct pyrrolidine ring, while RCM reaction was used to achieve the desired bicyclic framework. Comparing the two synthetic routes,it was found that amide 11 showed much better reactivity than amine 6 in the dihydroxylation reaction. Thus,the introduction of an electronwithdrawing group to the nitrogen atom was the highlight of the modified route. The synthesis of other difluoromethylenated castanospermine isomers as well as evaluation of their biological activity are currently on progress. Acknowledgments We thank the National Natural Science Foundation of China (Nos. 21272036,21332010) and the National Basic Research Program of China (No. 2012CB21600) for funding this work.
| [1] | (a) J.P. Michael, Indolizidine and quinolizidine alkaloids, Nat. Prod. Rep. 24 (2007) 191-222; (b) J.P. Michael, Indolizidine and quinolizidine alkaloids, Nat. Prod. Rep. 25 (2008) 139-165. |
| [2] | (a) I. Pastuszak, R.J. Molyneux, L.F. James, A.D. Elbein, Lentiginosine, a dihydroxyindolizidine alkaloid that inhibits amyloglucosidase, Biochemistry 29 (1990) 1886-1891; (b) A. Brandi, S. Cicchi, F.M. Cordero, et al., Assignment of the absolute configuration of natural lentiginosine by synthesis and enzymic assays of optically pure (+) and (-)-enantiomers, J. Org. Chem. 60 (1995) 6806-6812. |
| [3] | (a) M.J. Schneider, F.S. Ungemach, H.P. Broquist, T.M. Harris, (1S 2R,8R,8aR)-1,2,8-Trihydroxyoctahydroindolizine (swainsonine), an α-mannosidase inhibitor from Rhizoctonia leguminicola, Tetrahedron 39 (1983) 29-32; (b) G.P. Kaushal, T. Szumilo, I. Pastuszak, A.D. Elbein, Purification to homogeneity and properties of mannosidase II from mung bean seedlings, Biochemistry 29 (1990) 2168-2176. |
| [4] | (a) L.D. Hohenschutz, E.A. Bell, P.J. Jewess, et al., Castanospermine, A 1 6,7,8-tetrahydroxyoctahydroindolizine alkaloid, from seeds of Castanospermum austral, Phytochemistry 20 (1981) 811-814; (b) R.J. Nash, L.E. Fellows, J.V. Dring, et al., Castanospermine in Alexa species, Phytochemistry 27 (1988) 1403-1404. |
| [5] | (a) R. Saul, J.P. Chambers, R.J. Molyneux, A.D. Elbein, Castanospermine, a tetrahydroxylated alkaloid that inhibits β-glucosidase and β-glucocerebrosidase, Arch. Biochem. Biophys. 211 (1983) 593-597; (b) G. Trugnan, M. Rousset, A. Zweibaum, Castanospermine: a potent inhibitor of sucrase from the human enterocyte-like cell line Caco-2, FEBS Lett. 195 (1986) 28-32; (c) B.C. Campbell, R.J. Molyneux, K.C. Jones, Differential inhibition by castanospermine of various insect disaccharidases, J. Chem. Ecol. 13 (1987) 1759-1770; (d) A.M. Scofield, J.T. Rossiter, P. Witham, et al., Inhibition of thioglucosidasecatalysed glucosinolate hydrolysis by castanospermine and related alkaloids, Phytochemistry 29 (1990) 107-109; (e) A.P. Valaitis, D.F. Bowers, Purification and properties of the soluble midgut trehalase from the gypsy moth, Lymantria dispar, Insect Biochem. Mol. Biol. 23 (1993) 599-606. |
| [6] | (a) H. Nojima, I. Kimura, F.J. Chen, et al., Antihyperglycemic effects of N-containing sugars from Xanthocercis zambesiaca, Morus bombycis, Aglaonema treubii, and Castanospermum australe in Streptozotocin-Diabetic Mice, J. Nat. Prod. 61 (1998) 397-400; (b) R. Pili, J. Chang, R.A. Partis, et al., The α-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth, Cancer Res. 55 (1995) 2920-2926; (c) S.Walter, K. Fassbender, E. Gulbins, et al., Glycosylation processing inhibition by castanospermine prevents experimental autoimmune encephalomyelitis by interference with IL-2 receptor signal transduction, J. Neuroimmunol. 132 (2002) 1-10; (d) E. De Clercq, Current lead natural products for the chemotherapy of human immunodeficiency virus (HIV) infection, Med. Res. Rev. 20 (2000) 323-349; (e) P.M. Grochowicz, A.D. Hibberd, Y.C. Smart, et al., Castanospermine, an oligosaccharide processing inhibitor, reduces membrane expression of adhesion molecules and prolongs heart allograft survival in rats, Transpl. Immunol. 4 (1996) 275-285. |
| [7] | (a) T. Machan, A.S. Davis, B. Liawruangrath, S.G. Pyne, Synthesis of castanospermine, Tetrahedron 64 (2008) 2725-2732; (b) T. Jensen, M. Mikkelsen, A. Lauritsen, et al., A concise synthesis of castanospermine by the use of a transannular cyclization, J. Org. Chem. 74 (2009) 8886-8889; (c) J. Ceccon, G. Danoun, A.E. Greene, J.F. Poisson, Asymmetric synthesis of (+)-castanospermine through enol ether metathesis-hydroboration/oxidation, Org. Biomol. Chem. 7 (2009) 2029-2031; (d) G. Liu, T.J. Wu, Y.P. Ruan, P.Q. Huang, Asymmetric total syntheses of (+)-castanospermine, (+)-7-deoxy-6-epi-castanospermine, and (+)-1-epi-castanospermine, Chem, Eur. J. 16 (2010) 5755-5768; (e) E.G. Bowen, D.J. Wardrop, Diastereoselective nitrenium ion-mediated cyclofunctionalization: total synthesis of (+)-castanospermine, Org. Lett. 12 (2010) 5330-5333. |
| [8] | (a) J. Louvel, C. Botuha, F. Chemla, et al., Asymmetric total synthesis of (+)-6-epicastanospermine by the stereoselective formation of a syn, anti acetylenic 2-amino-1,3-diol stereotriad, Eur. J. Org. Chem. (2010) 2921-2926; (b) N.B. Kalamkar, V.G. Puranik, D.D. Dhavale, Synthesis of C1-and C8a-epimers of (+)-castanospermine from d-glucose derived g,d-epoxyazide: intramolecular 5-endo epoxide opening approach, Tetrahedron 67 (2011) 2773-2778; (c) P.R. Sultane, A.R. Mohite, R.G. Bhat, Total synthesis of 1-deoxy-7 8a-di-epicastanospermine and formal synthesis of pumiliotoxin-251D, Tetrahedron 53 (2012) 5856-5858; (d) H. Yun, J. Kim, J. Sim, et al., Asymmetric syntheses of 1-deoxy-6, 8a-di-epicastanospermine and 1-deoxy-6-epi-castanospermine, J. Org. Chem. 77 (2012) 5389-5393; (e) A.T. Serafidou, E.G. Yioti, J.K. Gallos, A protection-free synthetic access to (±)-1-deoxy-6-epi-castanospermine and (±)-1-deoxy-6 8a-di-epi-castanospermine, Eur. J. Org. Chem. (2013) 939-943. |
| [9] | R.H. Furneaux, G.J. Gainsford, J.M. Mason, et al., The chemistry of castanospermine, part V: synthetic modifications at C-1 and C-7, Tetrahedron 53 (1997) 245-268. |
| [10] | R.W. Wang, X.L. Qiu, M. Bols, O.C. Fernando, F.L. Qing, Synthesis and biological evaluation of glycosidase inhibitors: gem-difluoromethylenated nojirimycin analogues, J. Med. Chem. 49 (2006) 2989-2997. |
| [11] | (a) R.W. Wang, F.L. Qing, Highly stereocontrolled synthesis of gem-difluoromethylenated azasugars: D-and L-1,4,6-trideoxy-4,4-difluoronojirimycin, Org. Lett. 7 (2005) 2189-2192; (b) R.J. Li, M. Bols, C. Rousseau, et al., Synthesis and biological evaluation of potent glycosidase inhibitors: 4-deoxy-4,4-difluoroisofagomine and analogues, Tetrahedron 65 (2009) 3717-3727; (c) R.W. Wang, J. Xu, O. Lopez, M. Bols, F.L. Qing, Difluoromethylenated polyhydroxylated pyrrolidines: facile synthesis, crystal structure and biological evaluation, Future Med. Chem. 1 (2009) 991-997; (d) Y. Yang, F. Zheng, M. Bols, L.G. Marinescu, F.L. Qing, Synthesis of monofluorinated isofagomine analogues and evaluation as glycosidase inhibitors, J. Fluorine Chem. 132 (2011) 838-845. |
| [12] | G.E. Keck, M.B. Andrus, D.R. Romer, A useful new enantiomerically pure synthon from malic acid: chelation controlled activation as a route to regioselectivity, J. Org. Chem. 56 (1991) 417-420. |
| [13] | (a) H.C. Kolb, M.S. VanNieuwenhze, K.B. Sharpless, Catalytic asymmetric dihydroxylation, Chem. Rev. 94 (1994) 2483-2547; (b) Z.X. Jiang, Y.Y. Qin, F.L. Qing, Asymmetric synthesis of both enantiomers of anti-4,4,4-trifluorothreonine and 2-amino-4,4,4-trifluorobutanoic acid, J. Org. Chem. 68 (2003) 7544-7547. |
| [14] | S.G. Hentges, K.B. Sharpless, Asymmetric induction in the reaction of osmium tetroxide with olefins, J. Am. Chem. Soc. 102 (1980) 4263-4265. |
| [15] | K. Burgess, L.D. Jennings, Enantioselective esterifications of unsaturated alcohols mediated by a lipase prepared from Pseudomonas sp, J. Am. Chem. Soc. 113 (1991) 6129-6139. |
| [16] | (a) Z.W. You, Y.Y. Wu, F.L. Qing, Synthesis of gem-difluoromethylenated massoialactone by ring-closing metathesis, Tetrahedron Lett. 45 (2004) 9479-9481; (b) Z.W. You, X. Zhang, F.L. Qing, Stereocontrolled synthesis of gem-difluoromethylenated goniodiols and goniothalamin epoxides based on ring-closing metathesis, Synthesis (2006) 2535-2542. |