



 , Feng Qiana, Xiao-Man Wangc, Guan-Hai Tana, Rui-Xue Rongc, Zhi-Ran Caoc, Hua Chena,b, Ping-Zhu Zhanga,b, Xiao-Liu Lia,b
, Feng Qiana, Xiao-Man Wangc, Guan-Hai Tana, Rui-Xue Rongc, Zhi-Ran Caoc, Hua Chena,b, Ping-Zhu Zhanga,b, Xiao-Liu Lia,b
b Key Laboratory of Medicinal Chemistry and Molecular Diagnosis of Ministry of Education, Hebei University, Baoding 071002, China;
c Department of Immunology, School of Basic Medical Science, Hebei University, Baoding 071002, China
1. Introduction
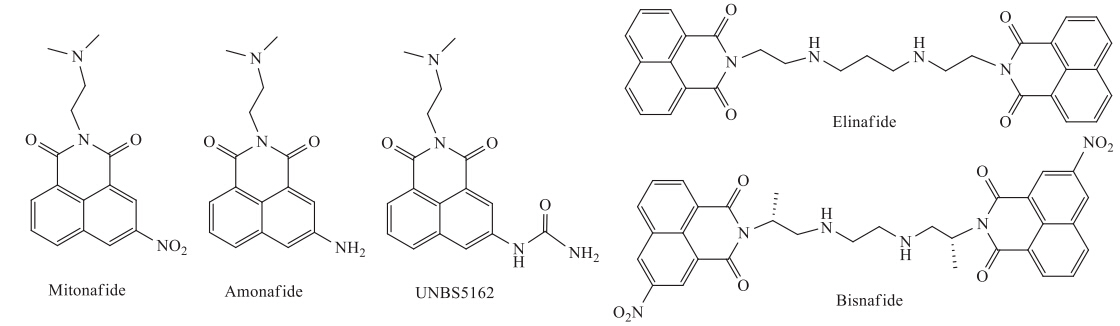
Naphthalimide derivatives,known to be DNA intercalators, have been widely investigated as anticancer agents for many years, and some compounds (Fig. 1) have reached clinical trials (Mitonafide,Amonafide,UNBS5162,Elinafide and Bisnafide) [1- 3]. Amonafide (Fig. 1) as the first compound reaching the clinical trial stage exhibited potent anticancer activity against solid tumors [1, 2, 3]. However,due to unexpected central neurotoxicity,hematotoxicity and limited efficiency,the further application of Amonafide in the clinical trial was limited [1, 2, 3]. In order to improve its selectivity,efficiency,and safety,a broad variety of novel naphthalimide derivatives have been developed [1, 2, 3, 4, 5, 6, 7, 8]. For example,UNBS5162,as a novel anticancer agent in a Phase I trial, showed potent anticancer activity and lower toxic side effects, which decreased CXCL chemokine expression in advanced solid tumors or lymphoma [4, 9]. Moreover,the synthesis of novel naphthalimide derivatives as potent anticancer agents is of considerable interest.

|
Download:
|
| Fig. 1. The structures of naphthalimide derivatives in clinical trials. | |
On the other hand,sodium dichloroacetate (DCA),as a targeting agent of the tumor metabolic process,has reached Phase II trials [10, 11, 12, 13]. Recently,the multifunctional drugs with DCA functionalization attracted special interest for their enhanced anticancer and lower toxic side effects [14]. For example,the multifunctional, hybrid,platinum (IV) prodrug with DCA functionalization [15, 16, 17], targeted to the metabolic process of the mitochondria and the DNA cross-linking process,showed much higher cytotoxicity toward the cancer cells,and paved a pathway for developing selective anticancer agents. In the interim,Cheng and coworkers reported a series of N-phenyl dichloroacetamide derivatives [18, 19, 20],which exhibited potent anticancer activity to cancer cells,and have valuable potential for drug development.
Inspired by the above results,novel naphthalimide derivatives, modified by dichloroacetamide conjugated with amino acid of different lengths (Scheme 1),were designed and synthesized in this paper. Their cytotoxic activities against human lung adenocarcinoma epithelial cells (A549),henrietta lacks strain of cancer cells (Hela) and human myelogenous leukemia cells (K562) were evaluated. Furthermore,their binding properties with calf-thymus DNA (Ct-DNA) were studied by UV-vis,fluorescence and circular dichroism spectroscopies and thermal denaturation experiment.

|
Download:
|
| Scheme 1. Reagents and conditions: (a) H2SO4,HNO3,5-20 ℃,82%; (b) N,N-dimethylethylenediamine,EtOH,reflux,82%; (c) 10% Pd/C,H2,EtOH,86%; (d) HATU,DIPEA,DMF; (e) CF3COOH,CH2Cl2; (f) dichloroacetyl chloride,TEA,CH2Cl2. | |
Melting points were measured with a SGW X-4 micro-melting point apparatus and were uncorrected. The 1H NMR,13C NMR spectra were recorded with RT-NMR Bruker AVANCE 600 NMR spectrometers. Chemical shifts were reported in ppm with TmS as the internal standard. High-resolution mass spectra (HRMS) were recorded with a Bruker Apex Ultra FTmS in the electrospray ionization (ESI) mode.
2.2. DNA-binding studiesThe UV-vis spectra and thermal denaturation studies were recorded on a Shimadzu UV-3600 spectrophotometer equipped with S-1700 temperature controller and Tm analysis soft. The fluorescent spectra were measured on a Hitachi F-7000 luminescence spectrophotometer. CD spectra were recorded on a Bio-Logic MOS-450/AF-CD. Ct-DNA was purchased from the Sigma-Aldrich Company. Solution of Ct-DNA in phosphate buffer (10 mmol L-1, pH 7.4) containing 50 mmol L-1 NaCl gave a ratio of UV absorbance at 260 and 280 nm of 1.8-1.9:1,indicating that the DNA was sufficiently free from protein. The concentration of Ct-DNA was determined by its absorption intensity at 260 nm with a known molar absorption coefficient value of 6600 mol-1 L cm-1.
2.3. Cytotoxicity analysisThe compounds 6a and 7a-d were dissolved in phosphate buffered saline (PBS) or PBS buffer containing 10% DMSO,and diluted to the required concentration with culture medium. The cytotoxicity was evaluated by MTT assay. Briefly,cells were plated in 96-well microassay culture plates (104 cells per well) and grown overnight at 37 ℃ in a 5% CO2 incubator. The compounds 6a and 7a-d were then added to the wells to achieve final concentrations ranging from10-7 to 10-4 mol L-1. The content of DMSO in the final solution of compounds 7a-d is less than 0.1%.Wells containing culturemedium without cells were used as control blanks; wells containing culture medium and cisplatin were used as positive control. The plates were incubated at 37 ℃ in a 5% CO2 incubator for 48 h. Upon completion of the incubation,stock 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma) dye solution (20 μL,5mg/μL) was added to each well. After 4 h incubation,2-propanol (100 μL) was added to solubilize theMTT formazan. The optical density of each well was then measured on a microplate spectrophotometer at a wavelength of 570 nm. The IC50 value was determined from the plot of % viability against dose of complexes added.
2.4. Synthesis 2.4.1. General synthesis of 5a-dBoc-protected amino acids (0.80 mmol) and 2-(7-aza-1Hbenzotriazole- 1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) (241 mg) were dissolved in 5 mL of dimethylformamide (DMF); the mixture was stirred at room temperature for 30 min,and then compound 4 (150 mg,0.53 mmol) and N,Ndiisopropylethylamine (DIPEA) (0.46 mL) were added. After that, the reaction mixture was stirred at room temperature for 18 h and then evaporated. The residue was dissolved with 100 mL of 5% hydrochloric acid solution. The solution was dried by Na2SO4,and evaporated in vacuo. The residues were further purified by silicagel column chromatography using CH2Cl2/MeOH (10:1,v/v),as the eluent,to give the product in the yields of 80% (5a),60% (5b),80% (5c) and 86% (5d),respectively.
Compound 5a:Mp 203.3-204.1 ℃; 1H NMR (600 MHz,CDCl3): δ 1.53 (s,9H,-CH3),2.44 (s,6H,-CH3),2.76 (s,2H,-CH2),4.07 (d,2H, J = 5.4 Hz,-CH2),4.34 (t,2H,J = 6.6 Hz,-CH2),5.58 (br,1H,-NH), 7.66 (t,1H,J = 7.2 Hz,Ar-H),8.03 (d,1H,J = 7.8 Hz,Ar-H),8.21 (s, 1H,Ar-H),8.41 (d,1H,J = 6.6 Hz,Ar-H),8.69 (s,1H,Ar-H),9.07 (br, 1H,CONH-Ar); 13C NMR (150 MHz,CDCl3): δ 28.36,37.96,45.67, 57.23,80.94,121.96,122.14,123.05,123.82,124.88,127.44, 129.88,132.31,133.75,136.37,163.69,164.15,168.63; HRMS (m/z): calcd. for C23H29N4O5+: 441.2138; found: 441.2126.
Compound 5b:Mp 205.7-206.9 ℃; 1H NMR (600 MHz,CDCl3): δ 1.46 (s,9H,-CH3),2.47 (s,6H,-CH3),2.72 (t,2H,J = 4.8 Hz,-CH2), 2.84 (br,2H,-CH2),3.57 (m,2H),4.34 (t,2H,J = 6.0 Hz,-CH2),5.30 (br,1H,-NH),7.62 (t,1H,J = 7.8 Hz,Ar-H),7.91 (d,1H,J = 6.6 Hz, Ar-H),8.06 (s,1H,Ar-H),8.33 (d,1H,J = 7.2 Hz,Ar-H),8.57 (s,1H, Ar-H),8.95 (s,1H,CONH-); 13C NMR (150 MHz,CDCl3): δ 28.45, 36.42,37.47,37.79,46.06,58.08,79.65,121.08,121.97,122.78, 123.16,124.37,127.37,129.54,132.15,133.64,136.79,156.32, 163.56,163.56,164.33,170.70; HRMS (m/z): calcd. for C24H31N4O5+: 455.2294; found: 455.2287.
Compound 5c:Mp 170.2-171.7 ℃; 1H NMR (600 MHz,CDCl3): δ 0.86 (m,2H),1.45 (s,6H,-CH3),1.48 (s,12H,-CH3),1.96 (t,2H, J = 6.0 Hz,-CH2),2.53 (m,2H,-CH2),2.89 (d,2H,J = 5.4 Hz,-CH2), 3.31 (d,2H,J = 5.4 Hz,-CH2),5.12 (s,1H,-NH),7.64 (t,1H, J = 7.8 Hz,Ar-H),8.04 (d,1H,J = 6.6 Hz,Ar-H),8.32 (s,1H,Ar-H), 8.36 (d,1H,J = 6.6 Hz,Ar-H),8.80 (s,1H,Ar-H),9.80 (s,1H, CONH-); 15C NMR NMR (150 MHz,CDCl3): δ 27.00,28.46,34.42,38.01, 39.34,45.90,57.51,79.97,121.58,122.12,122.92,123.97,124.64, 127.25,129.52,132.37,133.70,137.25,163.74,164.30,171.99; HRMS (m/z): calcd. for C25H33N4O5+: 469.2451; found 469.2443.
Compound 5d:Mp 187.1-188.7 ℃; 1H NMR (600 MHz,CD3OD): δ 1.43 (s,9H,-CH3),1.47 (m,2H,-CH2),1.57 (m,2H. -CH2),1.81 (m, 2H,-CH2),2.43 (s,6H,-CH3),2.49 (t,2H,J = 7.8 Hz,-CH2),2.76 (t, 2H,J = 7.2 Hz,-CH2),3.09 (t,2H,J = 6.6 Hz,-CH2),4.29 (t,2H, J = 6.6 Hz,-CH2),7.69 (t,1H,J = 7.8 Hz,Ar-H),8.12 (d,1H, J = 8.4 Hz,Ar-H),8.33 (d,1H,J = 7.2 Hz,Ar-H),8.46 (d,1H, J = 1.8 Hz,Ar-H),8.57 (s,1H,Ar-H); 13C NMR (150 MHz,CD3OD): δ 25.01,26.13,27.40,29.36,36.55,37.15,39.83,44.24,56.31,78.42, 121.56,121.84,122.59,123.95,124.46,127.05,129.11,132.26, 133.44,137.51,157.17,163.69,164.04,173.46; HRMS (m/z): calcd. for C27H37N4O5+: 497.2764; found 497.2756.
2.4.2. General synthesis of 6a-dIn a 25-mL flask,1.0 mmol each of compounds 5a-d was dissolved in 10 mL CH2Cl2. The solution was cooled to 0 ℃,and 10 mL of CF3COOH was carefully added under stirring. The mixture was allowed to warm to room temperature and stirred for 1 h. Then,the solvent was removed under reduced pressure. The residue was dissolved with 20 mL of 5% aqueous sodium carbonate. The organic phase was separated,washed with water, and dried over sodium sulfate. The product was obtained after the solvent was removed in vacuo. The yields of 6a were 90%,91%, 64% and 73%,respectively.
Compound 6a:Mp 118.5-119.9 ℃; 1H NMR (600 MHz,CD3OD): δ 3.06 (s,6H,-CH3),3.58 (t,2H,J = 5.4 Hz,-CH2),4.03 (s,2H,-CH2), 4.51(t,2H,J = 4.8 Hz,-CH2),7.70 (t,1H,J = 6.6 Hz,Ar-H),8.11 (d, 1H,J = 7.8 Hz,Ar-H),8.34 (d,1H,J = 6.6 Hz,Ar-H),8.44 (s,1H,Ar- H),8.50 (s,1H,Ar-H); 13C NMR (150 MHz,CD3OD): δ 35.16,41.05, 42.74,42.79,55.96,121.56,122.13,122.47,123.82,124.78,127.35, 129.89,132.26,133.92,136.74,164.05,164.37,165.00; HRMS (m/z): calcd. for C18H21N4O3+: 341.1613; found 341.1604.
Compound 6b: Mp 286.7-287.8 ℃ (hydrochloride); 1H NMR (600 MHz,CD3OD): δ 2.38 (s,6H,-CH3),2.70 (m,4H,-CH2),3.10 (t, 2H,J = 6.6 Hz,-CH2),4.33 (t,2H,J = 6.6 Hz,-CH2),7.76 (t,1H, J = 7.8 Hz,Ar-H),8.23 (d,1H,J = 7.8 Hz,Ar-H),8.44 (d,1H, J = 7.2 Hz,Ar-H),8.60 (d,1H,J = 1.8 Hz,Ar-H),8.71 (d,1H, J = 1.2 Hz,Ar-H); 13C NMR (150 MHz,CD3OD): δ 33.66,35.10, 35.70,43.22,46.05,55.31,121.60,122.42,123.25,124.36,124.72, 128.21,129.76,132.44,134.35,138.01,164.25,164.50,169.65; HRMS (m/z): calcd. for C19H23N4O3+: 355.1770; found 355.1762.
Compound 6c: Mp 72.6-73.5 ℃; 1H NMR (600 MHz,DMSO-d6): δ 2.81 (t,2H,J = 6.6 Hz,-CH2),2.90 (s,6H,-CH3),3.15 (m,2H,- CH2),3.32 (s,1H),3.45 (t,2H,J = 5.4 Hz,-CH2),4.38 (t,2H, J = 5.4 Hz,-CH2),7.85 (t,1H,J = 8.4 Hz,Ar-H),8.40 (m,2H,Ar-H), 8.71 (s,2H,Ar-H),the protons of another CH2 maybe hidden in the solvent peaks; 13C NMR (150 MHz,CD3OD): δ 22.74,33.12,35.11, 38.99,42.79,55.99,121.37,121.68,122.03,123.82,124.40,127.17, 129.54,132.12,133.83,137.38,164.08,164.35,171.92; HRMS (m/z): calcd. for C20H25N4O3+: 369.1927; found 369.1918.
Compound 6d:Mp 57.8-58.9 ℃; 1H NMR (600 MHz,DMSO-d6): δ 1.55 (m,2H,-CH2),1.77 (m,2H,-CH2),1.83 (m,2H,-CH2),2.55 (t, 2H,J = 7.2 Hz,-CH2),2.98 (t,2H,J = 7.8 Hz,-CH2),3.04 (s,6H,- CH3),3.55 (t,2H,J = 6.0 Hz,-CH2),4.56 (t,2H,J = 6.0 Hz,-CH2),7.82 (t,1H,J = 8.4 Hz,Ar-H),8.31 (d,1H,J = 8.4 Hz,Ar-H),8.52 (d,1H, J = 7.2 Hz,Ar-H),8.68 (d,1H,J = 1.8 Hz,Ar-H),8.78 (d,1H, J = 1.8 Hz,Ar-H); 13C NMR (150 MHz,CD3OD): δ 24.58,25.64, 26.96,35.24,36.13,39.20,42.84,56.13,115.81,117.75,121.65, 122.04,124.32,127.21,129.60,132.40,133.89,137.62,161.52, 161.75,164.25,164.52,173.32; HRMS (m/z): calcd. for C22H29N4O3+: 397.2240; found 397.2229.
2.4.3. General synthesis of 7a-dTo a solution of compounds 6a (1.0 mmol) and dichloroacetyl chloride (0.12 mL,1.3 mmol) in CH2Cl2 (10 mL) were added triethylamine (TEA) (0.3 mL). The reaction mixture was stirred at room temperature for 10 h and then evaporated. The residue was dissolved with 10 mL CH2Cl2,and washed with water. The solution was dried over Na2SO4,and evaporated in vacuo. The solids obtained were purified by column chromatography using CH2Cl2/MeOH (20:1,v/v) as the eluent to afford pure 7a (48%),7b (38%),7c (47%) and 7d (45%),respectively.
Compound 7a: Mp 186.2-189.3 ℃; 1H NMR (600 MHz,DMSOd6): δ 2.21 (s,6H,-CH3),2.52 (br,2H,-CH2),4.11 (s,2H,-CH2),4.15 (t,2H,J = 7.2 Hz,-CH2),6.65 (s,1H,-CH),7.82 (t,1H,J = 7.8 Hz,Ar- H),8.38 (t,2H,J = 7.2 Hz,Ar-H),8.63 (d,1H,J = 1.8 Hz,Ar-H),8.74 (d,1H,J = 1.8 Hz,Ar-H),9.02 (br,1H,CONH-),10.79 (br,1H, CONH-); 13C NMR (150 MHz,DMSO-d6): δ 35.69,43.22,43.70, 45.94,55.20,55.25,66.99,121.59,122.49,123.35,124.32,124.75, 128.15,129.67,132.53,134.37,138.08,164.49,164.43,164.70, 167.91; HRMS (m/z): calcd. for C20H21Cl2N4O4+: 451.0940; found 451.0936.
Compound 7b: Mp 169.1-165.7 ℃; 1H NMR (600 MHz,DMSOd6): δ 2.70 (t,2H,J = 6.6 Hz,-CH2),2.81 (s,3H,-CH3),3.48 (m,2H,- CH2),4.36 (t,2H,J = 5.4 Hz,-CH2),6.56 (s,1H,-CH),7.81 (t,1H, J = 7.8 Hz,Ar-H),8.37 (t,2H,J = 7.8 Hz,Ar-H),8.72 (s,1H,Ar-H), 8.77 (s,1H,Ar-H),8.96 (s,1H,CONH-),the protons of another CH2 maybe hidden in the solvent peaks; 13C NMR (150 MHz,CD3OD): δ 36.05,36.24,41.70,43.18,55.20,67.21,121.45,122.44,123.19, 124.50,124.66,128.02,129.49,132.48,134.27,138.41,164.18, 164.41,170.46; HRMS (m/z): calcd. for C21H23Cl2N4O4+: 465.1096; found 465.1087.
Compound 7c: Mp 174.5-176.3 ℃; 1H NMR (600 MHz,CD3OD): δ 1.83 (m,2H,-CH2),2.48 (m,2H,-CH2),2.87 (s,6H,-CH3),3.23 (m, 2H,-CH2),3.42 (s,2H,-CH2),4.39 (t,2H,J = 5.4 Hz,-CH2),6.59 (s, 1H,-CHCl2),7.82 (t,1H,J = 7.8 Hz,Ar-H),8.37 (t,2H,J = 8.4 Hz,Ar- H),8.72 (s,1H,Ar-H),8.79 (s,1H,Ar-H),8.96 (s,1H,CONH-),9.88 (s,1H,CONH-); 13C NMR (150 MHz,CD3OD): δ 24.99,34.06,35.60, 43.10,55.06,67.38,121.29,122.44,123.17,124.47,124.58,127.98, 129.38,132.52,134.29,138.57,164.13,134.19,134.43,172.07; HRMS (m/z): calcd. for C22H25Cl2N4O4+: 479.1252; found 479.1244.
Compound 7d: Mp 158.4-159.5 ℃; 1H NMR (600 MHz,DMSOd6): δ 1.35 (m,2H,-CH2),1.49 (m,2H,-CH2),1.67 (m,2H,-CH2), 2.43 (t,2H,J = 7.8 Hz,-CH2),3.13 (dd,2H,J = 6.6 Hz,12.6 Hz,-CH2), 3.42 (br,2H,-CH2),4.38 (t,2H,J = 6.0 Hz,-CH2) 6.51 (s,1H,-CH), 7.82 (t,1H,J = 7.8 Hz,Ar-H),8.37 (t,2H,J = 8.4 Hz,Ar-H),8.70 (d, 1H,J = 1.8 Hz,Ar-H),8.75 (br,1H,CONH-),8.79 (d,1H,J = 1.8 Hz, Ar-H),9.82 (br,1H,CONH-); 13C NMR (150 MHz,DMSO-d6): δ 25.12,26.33,28.76,34.56,36.79,43.14,45.88,55.14,67.41,121.32, 122.44,124.58,127.99,129.40,132.55,134.29,138.63,164.00, 164.23,134.46,172.59; HRMS (m/z): calcd. for C24H29Cl2N4O4+:507.1565; found 507.1559.
3. Results and discussionThe target compounds 7a-d were synthesized by six steps (Scheme 1). Commercially available 1,8-naphthalic anhydride 1 was reacted under concentrated H2SO4 and HNO3 conditions to afford compound 3-nitro-1,8-naphthalic anhydride 2 [21]. Then, compound 2 was condensed with N,N-dimethylethylenediamine to yield compound 3 [6],which was reduced by catalytic hydrogenation to obtain compound 4. Condensation of compound 4 with various Boc-protected amino acids yielded compounds 5a- d. Subsequent removal of the Boc protecting groups under TFA conditions,gave compounds 6a-d,which were directly condensed with dichloroacetyl chloride to form the corresponding target compounds 7a-d.
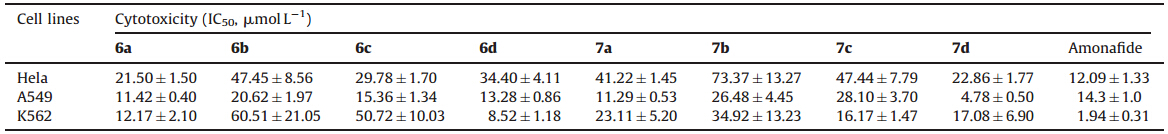
The cytotoxic activities of the naphthalimide derivatives 6a-d and 7a-d against cancer cell lines (Hela,A549 and K562) in vitro were measured with comparison to the control drug,Amonafide. As shown in Table 1,the length of the side chains of the amino acid influenced the cytotoxic activities. Compounds 6a with a glycine conjugate and 6d with an aminocaproic acid conjugate,and their dichloroacetamide derivatives 7a and 7d exhibited better cytotoxic activity than 6b,6c with b-alanine and 4-aminobutyric acid conjugates and their derivatives 7b and 7d. In addition,6a,6d,7a and 7d had more toxicity against A549 than against Hela and K562 cells compared with the control drug (Amonafide),especially 7d which had an IC50 value of 4.78 mmol L-1.
| Table 1 The cytotoxic activities of compounds 6a-d and 7a-d. |
Furthermore,the DNA binding properties of compounds 6a,6d and 7a,7d with Ct-DNA were investigated by using UV-vis, fluorescence and CD spectroscopies and DNA thermal denaturation experiment.
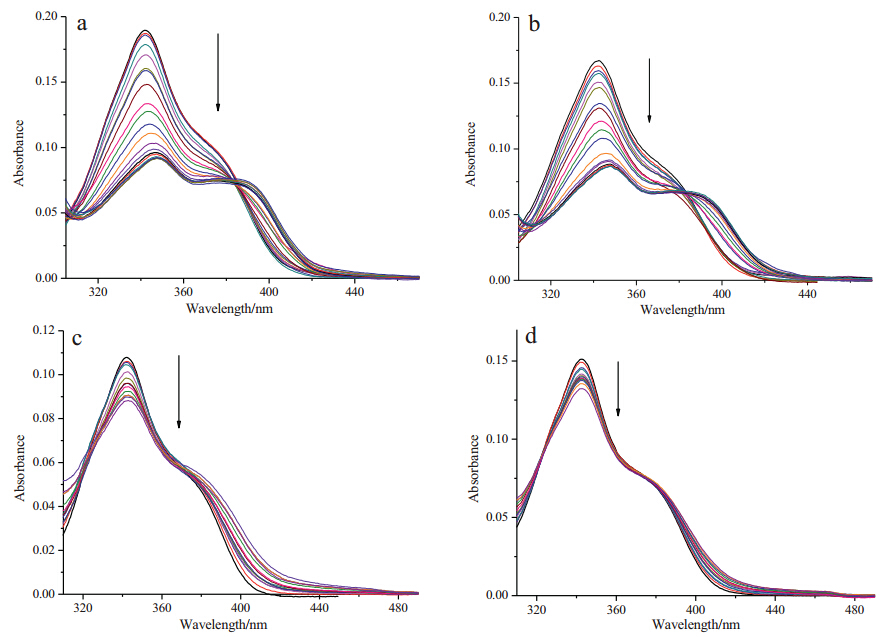
The UV-vis spectra of compounds 6a and 6d with Ct-DNA in PBS buffer (10 mmol L-1,pH 7.4,50 mmol L-1 NaCl) at 25 ℃ are shown in Fig. 2. The absorption intensity of compounds 6a and 6d decreased with the increase of Ct-DNA concentrations,and the maximum absorption peak showed a bathochromic shift of about 5 nm. This spectral characteristic implied that compounds 6a and 6d were inserted into the base pairs of DNA [22, 23, 24]. On the other hand,the DNA binding properties of the dichloroacetamide derivatives 7a and 7d with Ct-DNA showed hypochromicities upon addition of Ct-DNA,but the maximum absorption peak showed no obvious change. These results indicated that 7a and 7d exhibited weaker binding interactions with Ct-DNA than compounds 6a and 6d,as a result of the dichloroacetamide functionalization.

|
Download:
|
| Fig. 2. UV-vis spectral changes of compounds 6a and 6d (2.0 × 10-5 mol L-1,a and b),7a and 7d (2.0 × 10-5 mol L-1,c and d) upon addition of Ct-DNA (from 0 mol L-1 to 2 × 10-4 mol L-1) in phosphate buffer (10 mmol L-1,pH7.4,50 mmol L-1 NaCl) or in phosphate buffer (10 mmol L-1,pH7.4,50 mmol L-1 NaCl) containing 5% DMSO at 25 ℃. | |
The fluorescence spectra of compounds 6a,6d and 7a,7d with Ct- DNA are given in Fig. 3. Upon addition of Ct-DNA,the fluorescence of 6a,6d and 7a,7d was quenched. Furthermore,the binding constants between compounds 6a,6d and 7a,7d with Ct-DNA were obtained as 1.29 × 105 mol-1L (6a),1.15 × 105 mol-1L (6d), 5.1 × 103 mol-1L (7a),and 5.1 × 103 mol-1L (7d),respectively, using the nonlinear least-squares curve-fitting method [25] by fitting the experimental data of themaximumfluorescence changes.

|
Download:
|
| Fig. 3. Fluorescence spectral changes of compound 6a and 6d (2.0 × 10-5 mol L-1,a and b),7a and 7d (2.0 × 10-5 mol L-1,c and d) upon addition of Ct-DNA (from 0 mol L-1 to 2 × 10-4 mol L-1) in phosphate buffer (10 mmol L-1,pH7.4,50 mmol L-1 NaCl) or in phosphate buffer (10 mmol L-1,pH7.4,50 mmol L-1 NaCl) containing 5%DMSO at 25 ℃. | |
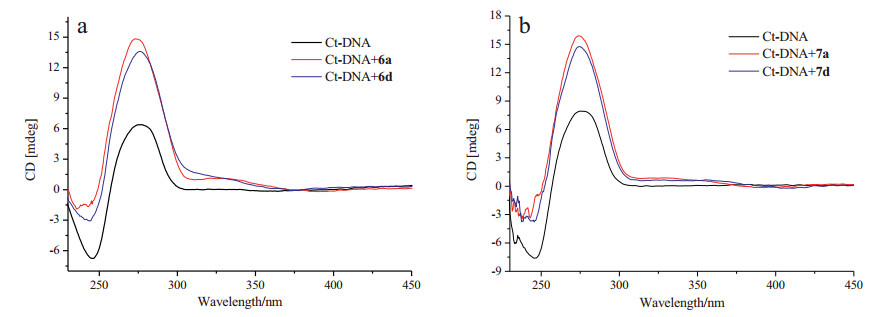
Circular dichroism (CD) is a powerful and reliable technique to investigate the conformational changes in DNA morphology during small molecules-DNA interactions. As shown in Fig. 4,the CD spectrum of free Ct-DNA showed a negative band at 246 nm due to the polynucleotide helicity,and a positive band at 276 nm due to the base staking,which conformed that the Ct-DNA existed in the right-band B form [26, 27]. Upon addition of compounds 6a,6d and 7a,7d,the CD signals of Ct-DNA underwent obvious changes in both the negative and positive bands without significant wavelength change. These results indicated that compounds 6a,6d and 7a,7d intercalated into the base pairs of DNA,causing the changes of the degree of rotation between successive base pairs [28, 29]. Additionally,weak,positive induced circular dichroism (ICD) signals were observed in the region of the characteristic absorption of the naphthalimides (300-380 nm),which indicated that 6a,6d and 7a,7d intercalated DNA with a vertical orientation in the intercalation pocket [30].

|
Download:
|
| Fig. 4. CD spectra of Ct-DNA (6.0 × 10-5 mol L-1) upon addition of 6a and 6d (1.0 × 10-4 mol L-1) in phosphate buffer (10 mmol L-1,pH7.4,containing 50 mmol L-1 NaCl) (a),and CD spectra of Ct-DNA (8.0 × 10-5 mol L-1) upon addition of 7a and 7d (1 × 10-4 mol L-1) in phosphate buffer (10 mmol L-1,pH 7.4,containing 50 mmol L-1 NaCl)containing 5% DMSO at 25 ℃ (b). | |
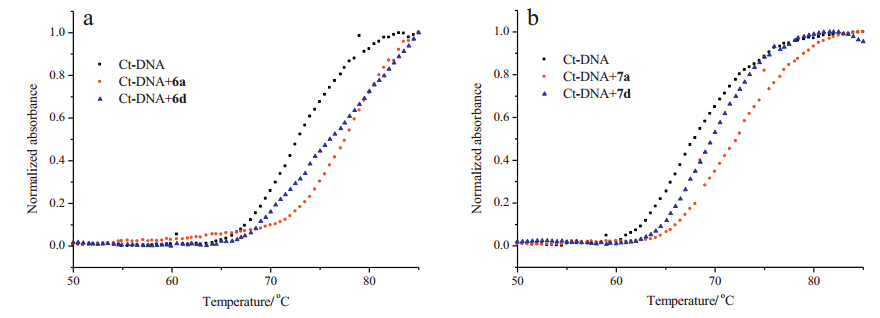
Moreover,the thermal denaturation experiments of the Ct-DNA in the absence,and presence,of 6a,6d and 7a,7d (Fig. 5) showed an obvious change in the melting temperatures (Tm),which could provide useful information on the conformational change and the interaction strength between the DNA and the compounds [31].

|
Download:
|
| Fig. 5. DNA melting curves for Ct-DNA (5.0 × 10-5 mol L-1) in the absence and presence of 6a and 6d (5.0 × 10-6 mol L-1) in phosphate buffer (1 mmol L-1,pH 7.4,5 mmol L-1 NaCl) (a),7a and 7d (5.0 × 10-6 mol L-1) in phosphate buffer (1 mmol L-1,pH 7.4,5 mmol L-1 NaCl) containing 5% DMSO (b). | |
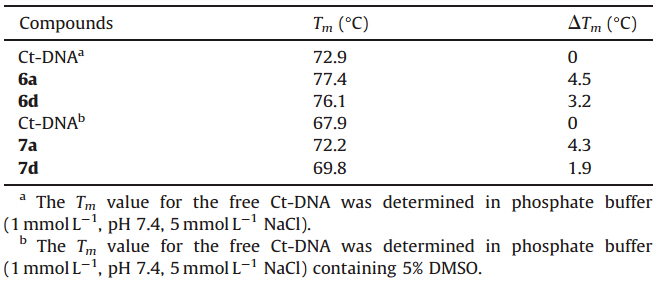
The melting curves of Ct-DNA are illustrated in Fig. 5 and Table 2,respectively. The Tm value for the free Ct-DNA is 72.9 ℃ in PBS buffer. Upon addition of 6a,6d,obvious changes in the DNA melting temperature were observed and the Tm values increased to 77.4 ℃ and 76.1 ℃,respectively. The increases of the melting temperature (ΔTm) induced by DNA-compound interactions were 4.5 ℃ and 3.2 ℃,respectively,indicating that compound 6d possessed a lower DNA melting temperature than 6a. On the other hand,the Tm value for the free Ct-DNA in PBS buffer containing 5% DMSO was 67.9 ℃. Upon addition of compounds 7a,and 7d,the increases in the melting temperature (ΔTm) induced by DNA- compound interactions were 4.3 ℃,and 1.9 ℃,respectively,which suggested that compound 7d had a lower DNA melting temperature than 7a. These results illustrated that glycine conjugated naphthalimide derivatives 6a and 7a showed the stronger binding interactions with the Ct-DNA than the aminocaproic acid linked derivatives 6d and 7d,which was consistent with the results from the fluorescence titration data.
| Table 2 Average Tm and DTm for Ct-DNA in the absence and presence of 6a,6d and 7a,7d. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Considering the above results,we could conclude that the novel naphthalimides derivatives 6a and 7a-d,as the DNA intercalators, possessed similar binding properties with DNA. The cytotoxic activity and the binding property with DNA of the amino acids modified naphthalimide derivatives and their dichloroacetamide functionalized derivatives showed a similar trend with the side chains.
4. ConclusionA series of novel naphthalimide derivatives modified by amino acids and dichloroacetamide at 3-position have been synthesized. Their cytotoxic activities were preliminarily evaluated against Hela,A549 and K562 cells and their DNA binding properties were investigated by UV-vis,fluorescence,and circular dichroism (CD) spectroscopies and thermal denaturation experiment. The results revealed that the lengths of the side chain of the amino acids influenced the cytotoxic activities and the binding properties. The glycine and aminocaproic acid modified naphthalimide derivatives 6a (7a) and 6d (7d) exhibited better cytotoxic activity. Especially, compound 7d with an IC50 value of 4.78 μmol L-1 against A549 cells would dictate further investigation to develop a potential anticancer agent.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21372059 and 21172051),the Hebei Natural Science Foundation (No. B2012201041) and the Foundation of Hebei Education DeparTment (No. YQ2013006).
| [1] | M.F. Braña, A. Ramos, Naphthalimides as anti-cancer agents: synthesis and biological activity, Curr. Med. Chem. -Anticancer Agents 1 (2001) 237-255. |
| [2] | L. Ingrassia, F. Lefranc, R. Kiss, T. Mijatovic, Naphthalimides and azonafides as promising anti-cancer agents, Curr. Med. Chem. 16 (2009) 1192-1213. |
| [3] | M. Lv, H. Xu, Overview of naphthalimide analogs as anticancer agents, Curr. Med. Chem. 16 (2009) 4797-4813. |
| [4] | T. Mijatovic, T. Mahieu, C. Bruyère, et al., UNBS5162, a novel naphthalimide that decreases CXCL chemokine expression in experimental prostate cancers, Neoplasia 10 (2008) 573-586. |
| [5] | E.V. Quaquebeke, T. Mahieu, P. Dumont, et al., 2,2,2-Trichloro-N-({2-[2-(dimethylamino) ethyl]-1,3-dioxo-2,3-dihydro-1H-benzo[de]isoquinolin-5-yl}carbamoyl) acetamide (UNBS3157), a novel nonhematotoxic naphthalimide derivative with potent antitumor activity, J. Med. Chem. 50 (2007) 4122-4134. |
| [6] | A. Kamal, N.R. Bolla, P.S. Srikanth, A.K. Srivastava, Naphthalimide derivatives with therapeutic characteristics: a patent review, Expert. Opin. Ther. Patents 23 (2013) 299-317. |
| [7] | S. Banerjee, E.B. Veale, C.M. Phelan, et al., Recent advances in the development of 1,8-naphthalimide based DNA targeting binders, anticancer and fluorescent cellular imaging agents, Chem. Soc. Rev. 42 (2013) 1601-1618. |
| [8] | S.Y. Tan, H. Yin, Z. Chen, X.H. Qian, Y.F. Xu, Oxo-heterocyclic fused naphthalimides as antitumor agents: synthesis and biological evaluation, Eur. J. Med. Chem. 62 (2013) 130-138. |
| [9] | D. Mahadevan, D.W. Northfelt, P. Chalasani, et al., Phase I trial of UNBS5162, a novel naphthalimide in patients with advanced solid tumors or lymphoma, Int. J. Clin. Oncol. 18 (2013) 934-941. |
| [10] | D.A. Tennant, R.V. Duraán, E. Gottlieb, Targeting metabolic transformation for cancer therapy, Nat. Rev. Cancer 10 (2010) 267-277. |
| [11] | I. Papandreou, T. Goliasova, N.C. Denko, Anticancer drugs that target metabolism: is dichloroacetate the new paradigm? Int. J. Cancer 128 (2011) 1001-1008. |
| [12] | C. Granchi, F. Minutolo, Anticancer agents that counteract tumor glycolysis, ChemMedChem 7 (2012) 1318-1350. |
| [13] | F. Wang, M.A. Ogasawara, P. Huang, Small mitochondria-targeting molecules as anti-cancer agents, Mol. Aspects Med. 31 (2010) 75-92. |
| [14] | J.S. Butler, P.J. Sadler, Targeted delivery of platinum-based anticancer complexes, Curr. Opin. Chem. Biol. 17 (2013) 175-188. |
| [15] | S. Dhar, S.J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 22199-22204. |
| [16] | H.H. Xiao, L.S. Yan, Y. Zhang, et al., A dual-targeting hybrid platinum(IV) prodrug for enhancing efficacy, Chem. Commun. 48 (2012) 10730-10732. |
| [17] | X.Xue, S.You,Q.Zhang, et al.,Mitaplatinincreases sensitivity of tumor cells tocisplatin by inducing mitochondrial dysfunction, Mol. Pharmacol. 9 (2012) 634-644. |
| [18] | Y.C. Yang, P.H. Shang, C.M. Cheng, et al., Novel N-phenyl dichloroacetamide derivatives as anticancer reagents: design, synthesis and biological evaluation, Eur. J. Med. Chem. 45 (2010) 4300-4306. |
| [19] | T.W. Li, Y.C. Yang, C.M. Cheng, et al., Design, synthesis and biological evaluation of N-arylphenyl-2,2-dichloroacetamide analogues as anti-cancer agents, Bioorg. Med. Chem. Lett. 22 (2012) 7268-7271. |
| [20] | T.W. Li, Y.C. Yong, C.M. Cheng, et al., Multi-substituted N-phenyl-2,2-dichloroacetamide analogues as anti-cancer drugs: design, synthesis and biological evaluation, Acta Pharm. Sin. 47 (2012) 354-363. |
| [21] | J.A. Montgomery, A.T. Shortnacy, D.A. Carson, J.A. Secrist III, Synthesis and biological evaluation of irreversible inhibitors of aldose reductase, J. Med. Chem. 29 (1986) 2384-2389. |
| [22] | E.C. Long, J.K. Barton, On demonstrating DNA intercalation, Acc. Chem. Res. 23 (1990) 271-273. |
| [23] | X. Yang, W. Liu, W. Jin, G. Shen, R. Yu, DNA binding studies of a solvatochromic fluorescence probe 3-methoxybenzanthrone, Spectrochim. Acta A 55 (1999) 2719-2727. |
| [24] | L.J. Xie, Y.F. Xu, F. Wang, et al., Synthesis of new amonafide analogues via coupling reaction and their cytotoxic evaluation and DNA-binding studies, Bioorg. Med. Chem. 17 (2009) 804-810. |
| [25] | M. Kožurkovaá, D. Sabolovaá, H. Paulíkovaá, et al., DNA binding properties and evaluation of cytotoxic activity of 9,10-bis-N-substituted (aminomethyl)anthracenes, Int. J. Biol. Macromol. 41 (2007) 415-422. |
| [26] | P. Uma Maheswari, M. Palaniandavar, DNA binding and cleavage activity of[Ru(NH3)4(diimide)]Cl2 complexes, Inorg. Chim. Acta 357 (2004) 901-912. |
| [27] | X. Jiang, L. Shang, Z.X. Wang, S.J. Dong, Spectrometric and voltammetric investigation of interaction of neutral red with calf thymus DNA: pH effect, Biophys. Chem. 118 (2005) 42-50. |
| [28] | P.U. Maheswari, M. Palaniandavar, DNA binding and cleavage properties of certain tetrammine ruthenium(II) complexes of modified 1,10-phenanthrolines: effect of hydrogen-bonding on DNA-binding affinity, J. Inorg. Biochem. 98 (2004) 219-230. |
| [29] | X.L. Li, Y.J. Lin, Q.Q. Wang, et al., The novel anti-tumor agents of 4-triazol-1,8-naphthalimides: synthesis, cytotoxicity, DNA intercalation and photocleavage, Eur. J. Med. Chem. 46 (2011) 1274-1279. |
| [30] | Z.C. Zhang, Y.Y. Yang, D.N. Zhang, et al., Acenaphtho[1,2-b]pyrrole derivatives as new family of intercalators: various DNA binding geometry and interesting antitumor capacity, Bioorg. Med. Chem. 14 (2006) 6962-6970. |
| [31] | U. Chaveerach, A. Meenongwa, Y. Trongpanich, C. Soikum, P. Chaveerach, DNA binding and cleavage behaviors of copper(II) complexes with amidino-O-methylurea and N-methylphenyl-amidino-O-methylurea, and their antibacterial activities, Polyhedron 29 (2010) 731-738. |