




Amorphous calcium phosphates (ACPs),a kind of calcium phosphate ceramics,are present in the early stage of bone biomineralization and eventually convert to stable crystalline hydroxyapatites (HAs) [1, 2, 3]. ACP has demonstrated better osteoconductivity [4] and crack resistance than hydroxyapatite (HA) [5],and is more biodegradable than tricalcium phosphate (TCP) [6]. Another key feature of the ACP nanoparticles is its intrinsic porosity that is useful for loading proteins,genes,siRNA and drugs [7, 8, 9, 10, 11].
From a thermodynamic point of view,amorphous materials are at best metastable [12]. ACP precipitates should be collected and freeze-dried shortly after the preparation (the sooner,the better), because in aqueous or non-aqueous media ACP is spontaneously converted to other crystalline calcium orthophosphates,mainly calcium deficient hydroxyapatite (CDHA) [13]. It is widely accepted that the dissolution of ACP and the nucleation and growth of HA from solution are responsible for the phase transformation processes. Organic solvents or organic additives were widely used to stabilize ACP because the complexes of calcium with organic agents can form during the synthesis. This favors the ACP formation,which is attributed to the coordinated complexing agents remaining in the structure of ACP. However, amorphous calcium phosphate (ACP) synthesized with cyclodextrins (CDs) [14],polyethylene glycol (PEG) [15],terephthaloyl chloride [16] or ethylene glycol (EG) [17] as organic additives at or below room temperature in aqueous solution could remain stable in aqueous solution for several days.
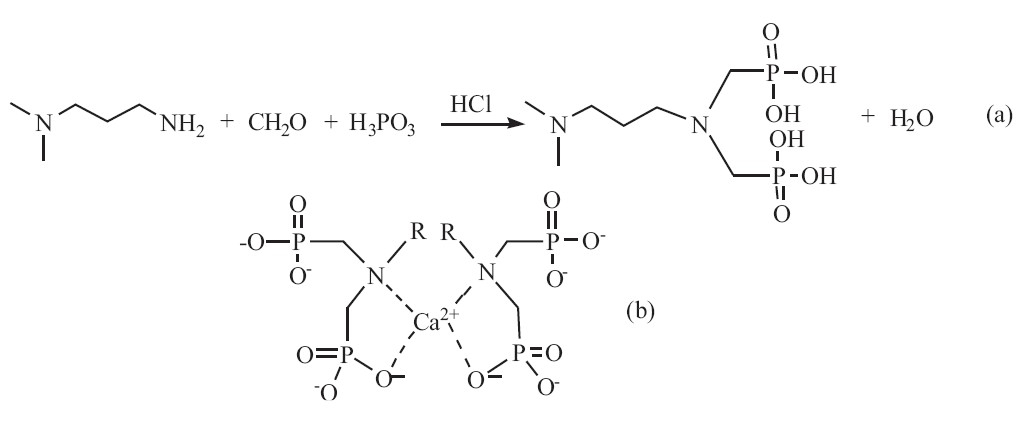
We have synthesized a novel surface modifier for HA,i.e., a-methoxy-v-dihydrogen phosphate-poly (ethylene glycol) (mPEG-OPO3H2). Its organic phosphate groups could strongly bond to Ca2+ ions of the growing HA particles during their synthesis in water,forming the HA nanoparticles with peripheral methoxy-poly (ethylene glycol) (mPEG) chains [18]. Bisphosphates (BPs) show higher affinity to calcium phosphate than their mono-phosphate counterparts and are widely used for the treatment of orthopedic diseases,such as Paget bone disease,osteoporosis,fibrous dysplasia, myeloma and bone metastases [19, 20, 21, 22]. Based on this,we proposed that organic bisphosphates could be used to stabilize ACP for a longer time by being tightly bonded on the surface of ACP and provide the ACP particles with good redispersibility and colloid stability. Therefore,we synthesized a bisphosphate,i.e. 3-(N,Ndimethylamino)- N',N'-bis(phosphonomethyl) propylamine (NDBP), a biocompatible aminophosphonic acid [23]. So we prepared ACP by using NDBP as an organic phosphate and surface modifier. The synthetic route ofNDBP andthe supposed complexationmechanism of NDBP with calcium ions [24] are shown in Scheme 1.

|
Download:
|
| Scheme 1.(a) The synthetic route of NDBP; (b) complexation of calcium ion by BPs. | |
{kind=link}
Aqueous ammonia (25% by mass fraction),concentrated hydrochloric acid and formaldehyde solution (30-40% by mass fraction) and Ca(NO3)2·4H2O,(NH4)2HPO4 and H3PO3 were all of analytical grade and purchased from Chengdu Kelong Chemical Reagent Company,Chengdu,Sichuan,China. 3-Dimethylaminopropylamine (C5H14N2) was purchased from Adamas Reagent Co., Ltd.,Shanghai,China. 2.2. Synthesis of 3-(N,N-dimethylamino)-N',N'-bis(phosphonomethyl)propylamine (NDBP)
NDBP was prepared using the Mannich reaction [25]. Briefly,3- dimethylaminopropylamine (1.02 g,0.01 mol),H3PO3 (1.64 g, 0.02 mol) and a stirring bar were added to a three-necked round-bottom flask,and deionized water (28 mL) was added. The flask was placed in an ice-salt-bath and concentrated hydrochloric acid (5.4 mL) was slowly added to the flask drop wise. After heating to 90 ℃,formaldehyde solution (5.4 mL) was added drop wise,and the reaction mixture was kept at reflux temperature for 3 hours. The solvent was removed using a rotary evaporator and the product was washed with anhydrous ethanol and stored in a dry environment. The product was characterized by NMR and FTIR. 1H NMR in D2O gave the following resonances: δ 2.15 (m,2H,C-CH2-C),2.79-3.12 (s,6H,-D3O),3.22-3.26 (d,4H, N-CH2-P),3.41-3.45 (m,4H,N-CH2-C); 13C NMR (D2O): δ 54.09(d, N-CH2-P),53.14 (-CH2-N),51.78 (N-CH2-),42.81 (-D3O), 19.13(C-CH2-C); FTIR (cm-1): 3435.48 (νP-OH),1473.83 (νP=O), 923.32 (νP-O). 2.3. Synthesis of amorphous calcium phosphates (ACPs)
ACP nanoparticles were synthesized with NDBP as a surface modifier by using the similar synthetic procedures that we have reported recently [18]. The phosphate solution was prepared by dissolving 0.0609 g of NDBP (organic phosphate 0.41 mmol) and 0.0238 g of (NH4)2HPO4,(inorganic phosphate,0.18 mmol) in 16 mL of water. The calcium solution was prepared by dissolving 0.236 g of Ca(NO3)2·4H2O (1 mmol) in 14 mL of water. The total Ca/P molar ratio was 1.67. The pH values of both solutions were adjusted to 10.5 by aqueous ammonia. For the preparation of ACP,the calcium solution was added drop wise into the phosphate solution at room temperature in 15 min,followed by ultrasonic irradiation for 30 min,during which the pH value of themixture was kept at 10.5. Thereafter,themixture was aged at 85 ℃ for 5 h. The resulting light blue hydrocolloid was subjected to 30 min ultrasonic treatment and then stayed overnight before purification.
The colloid was centrifuged at 15,000 rpm,followed by water washing and re-dispersing repeatedly until the conductivity of the re-dispersed colloid was near that of water,indicating that water soluble impurities were totally removed. Most centrifuged precipitate was freeze-dried for 24 h to obtain powders. The residue was added to water or methanol and then subjected to 15 min of ultrasonication irradiation to form a stable colloid. The controlled sample was prepared using the same synthesis as mentioned above but without any surface modifiers and designated as BLK.
In order to investigate the long term stability of the ACP particles in water,the re-dispersed ACP aqueous colloids were stored at room temperature for 2,4 and 6 months,respectively, before being centrifuged and freeze-dried. The samples were designated as ACP-2m,ACP-4m and ACP-6m,respectively. The freeze-dried powders of ACP-2m,ACP-4m and ACP-6m were subjected for the XRD study. 2.4. Characterization
The X-ray diffraction (XRD) patterns of the ACP and BLK powders were recorded on an X’Pert Pro MPD diffractometer (PANalytical BV,Netherlands) using a Cu Kα radiation operated at 40 (kV) and 35 (mA). Fourier transform infrared (FTIR,KBr pellets) was carried out on a Nicolet IR200 spectrometer (Thermo Electron, USA),in the wavenumber range 400-4000 cm-1 with a resolution of 4 cm-1. The Ca/P ratio of the ACP was measured by Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES) (ARCOS type of SPECTRO Analytical Instruments GmbH in Germany). The particle size and size distribution of ACP-0m were measured using a Mastersier 2000 (Malvern Instruments,UK) dynamic light scattering (DLS) (Malvern ZEN3690,UK). The transmission electron microscopy (TEM) micrographs were recorded for the colloidal appetites on a JEOL JEM-100CX microscope (Japan),set at an accelerating voltage of 80 kV. 3. Results and discussion
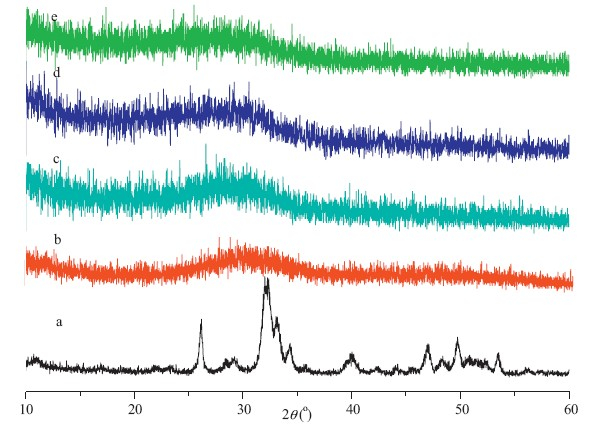
The XRD patterns of BLK and as-prepared ACP (ACP-0m) nanoparticles are shown in Fig. 1a and Fig. 1b,respectively. From Fig. 1a,we can see that the BLK sample demonstrated some weak diffraction peaks that match those of standard HA (JCPDS NO. 9- 0342),indicating their poorly crystalline HA structure. On the other hand,a single broad peak was observed at around 2θ= 32° for sample of ACP-0m,revealing their amorphous nature. The ICP-AES test showed that the Ca/P molar ratio of ACP-0m powder was 1.64. Considering the XRD results,we can conclude that the ACP-0m particles were amorphous calcium phosphate.

|
Download:
|
| Fig. 1.XRD patterns of ACP dried powder (a,BLK; b,ACP-0m; c,ACP-2m; d,ACP-4m;e,ACP-6m). | |
{kind=link}
The XRD patterns of the ACP powders centrifuged and freezedried from ACP aqueous colloids that were stored at room temperature for 2 months (ACP-2m),4 months (ACP-4m) and 6 months (ACP-6m) are shown in Fig. 1c,d and e,respectively. It can be seen from Fig. 1c,d and e that a single broad peak centered at around 2θ = 32° was observed for all three samples,suggesting their amorphous nature. Therefore,we can conclude that the ACP nanoparticles retained amorphous structure even after being stored for 6 months in water. However,the ACP nanoparticles prepared by using PEG,CD,terephthaloyl chloride and EG as additives at room temperature retained their amorphous structure for just several days at most as reported in published literatures [14, 15, 16, 17]. Compared with these additives,the phosphate group and tertiary amine group in our additive were adsorbed on the surface of the ACP particles by forming more stable coordinated complexes with calcium ion as shown in Scheme 1b. The organic bisphosphate molecules adsorbed on the surface of the ACP particles prevented the ACP from transforming to crystallized structures by protecting the ACP particles from dissolution. The adsorbed organic bisphosphate molecules also rendered the ACP nanoparticles with good redispersibility and stabilized the ACP hydrocolloid for more than 6 months.
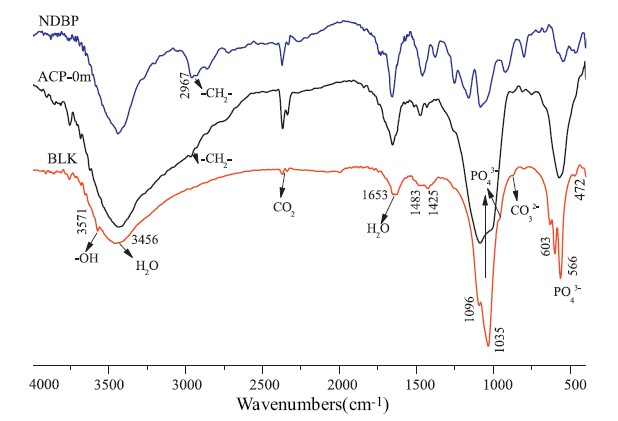
The FTIR spectra of ACP-0m,BLK and NDBP are shown in Fig. 2. For BLK samples,the obvious phosphate stretching bands at 1096, 1033 and 959 cm-1 and the well-splitting bending bands at 603, 504 and 474 cm-1 proved the structurally ordered (crystal) environment of the PO43- groups [26, 27],which are in agreement with the XRD results. The absorption band at 3570 cm-1 in the sample of BLK arises from the hydroxyl group,which further proves the identity of the BLK sample as HA. However,the adsorption band of hydroxyl group at 3571 cm-1 disappeared and the bending vibrations of phosphate in 400-600 cm-1 merged into a broad single peak for sample of ACP-0m,suggesting its amorphous structure. The -CH2- stretching band at ~2958 cm-1 in ACP-0m revealed the presence of the NDBP despite the fact that the sample was thoroughly washed before the FTIR characterization. In contrast,no adsorption bands at ~2900 cm-1 were found in the FTIR spectra for BLK sample. The broad adsorption at 3450 cm-1 in all samples was an indication of adsorbed water. So,we suggested that the phosphate groups in NDBP anchored on the ACP particles by the strong interactions with the calcium ions in the ACP particles,which prevented ACP from transforming into HA by the dissolution and nucleation mechanism. The organic chains that anchored on the ACP particles were also responsible for their redispersibility and long colloid stability in water.

|
Download:
|
| Fig. 2.FTIR spectra of ACP-0m,BLK and NDBP. | |
{kind=link}
The size and size distribution of ACP-0m hydrocolloid tested by DLS was shown Fig. 3a,which demonstrated fairly narrow particle size distributions and an average hydrodynamic size of 108 nm. The ACP-0m hydrocolloid remained stable for over 6 months,which further confirmed the tightly bonded NDBP chains onto each ACP particle as indicated by FTIR. Otherwise the suspending HA particles precipitated rapidly in several minutes for BLK sample due to the lack of surface modification as proved by FTIR.

|
Download:
|
| Fig. 3.Particle size distribution and TEM image of ACP-0m hydrocolloid (a,particle size distribution of ACP-0m; b,TEM image of ACP-0m). | |
{kind=link}
The morphology of the ACP-0m hydrocolloid characterized by TEM was shown in Fig. 3b. It can be seen from Fig. 3b that the ACP particles presented spherical morphology without severe agglomeration and the particles size was about 60 nm,which is smaller than the DLS results ascribed to the dehydration of the ACP particles during TEM observation [28]. In fact,ACP nanoparticles usually show curvilinear appearance rather than the faceted and angular shape of crystalline calcium orthophosphates as reported previously [29, 30, 31],which is consistent with our results. 4. Conclusion
We have synthesized a new bisphosphate,i.e. 3-(N,N-dimethylamino)- N',N'-bis(phosphonomethyl) propylamine (NDBP),and prepared ACP using NDBP as an organic phosphate and surface modifier. The organic bisphosphates decreased the crystallinity and prevented the ACP from transforming to crystalline HA and stabilized the ACP nanoparticles in water for more than 6 months. The surface modifier tightly bonded onto the ACP nanoparticles also significantly enhanced the redispersibility and colloidal stability of the ACP nanoparticles. Acknowledgments
We acknowledge the financial supports from the Natural Science Foundation of China (No. 50973069),the project of Postgraduate Degree Construction,Southwest University for Nationalities (No. 2013XWD-S0703).
| [1] | H.A. Lowenstam, S. Weiner, Transformation of amorphous calcium phosphate to crystalline dahillite in the radular teeth of chitons, Science 227 (1985) 51-53. |
| [2] | I.M. Weiss, N. Tuross, L. Addadi, S. Weiner, Mollusc larval shell formation: amorphous calcium carbonate is a precursor phase for aragonite, J. Exp. Zool. 293 (2002) 478-491. |
| [3] | E. Beniash, J. Aizenberg, L. Addadi, S. Weiner, Amorphous calcium carbonate transforms into calcite during sea urchin larval spicule growth, Proc. R. Soc. B: Biol. Sci. 264 (1997) 461-465. |
| [4] | M. Nagano, T. Nakamura, T. Kokubo, M. Tanahashi, M. Ogawa, Differences of bone bonding ability and degradation behaviour in vivo between amorphous calcium phosphate and highly crystalline hydroxyapatite coating, Biomaterials 17 (1996) 1771-1777. |
| [5] | A.S. Posner, F. Betts, Synthetic amorphous calcium phosphate and its relation to bone mineral structure, Acc. Chem. Res. 8 (1975) 273-281. |
| [6] | T. Kαnazawa, T. Umegaki, N. Uchiyama, Thermal crystallisation of amorphous calcium phosphate to a-tricalcium phosphate, J. Chem. Technol. Biotechnol. 32 (1982) 399-406. |
| [7] | E.T. Hwang, R. Tatavarty, J.Y. Chung, M.B. Gu, New functional amorphous calcium phosphate nanocomposites by enzyme-assisted biomineralization, ACS Appl. Mater. Interfaces 5 (2013) 532-537. |
| [8] | J. Li, Y.C. Chen, Y.C. Tseng, S. Mozumdar, L. Huang, Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery, J. Control. Release 142 (2010) 416-421. |
| [9] | A. Oyane, H. Araki, Y. Sogo, A. Ito, H. Tsurushima, Spontaneous assembly of DNA- amorphous calcium phosphate nanocomposite spheres for surface-mediated gene transfer, CrystEngComm-15 (2013) 4994-4997. |
| [10] | M. Epple, K. Ganesan, R. Heumann, et al., Application of calcium phosphate nanoparticles in biomedicine, J. Mater. Chem. 20 (2010) 18-23. |
| [11] | C. Qi, Y.J. Zhu, F. Chen, Fructose 1,6-bisphosphate trisodium salt as a new phosphorus source for the rapid microwave synthesis of porous calcium-phosphate microspheres and their application in drug delivery, Chem. Asian J. 8 (2013) 88-94. |
| [12] | P. Keblinski, S.R. Phillpot, D. Wolf, H. Gleiter, Thermodynamic criterion for the stability of amorphous intergranular films in covalent materials, Phys. Rev. Lett. 77 (1996) 2965-2968. |
| [13] | J. Christoffersen, M.R. Christoffersen, W. Kibalczyc, F.A. Andersen, A contribution to the understanding of the formation of calcium phosphates, J. Cryst. Growth 94 (1989) 767-777. |
| [14] | Y.B. Li, T. Wiliana, K.C. Tam, Synthesis of amorphous calcium phosphate using various types of cyclodextrins, Mater. Res. Bull. 42 (2007) 820-827. |
| [15] | C.F. Qiu, X.F. Xiao, R.F. Liu, Biomimetic synthesis of spherical nano-hydroxyapatite in the presence of polyethylene glycol, Ceram. Int. 34 (2008) 1747-1751. |
| [16] | R. Li, G.M. Chen, X.L. Ma, et al., Mineralization of HA crystals regulated by terephthaloyl chloride-modified silk fibroin films, Chin. Chem. Lett. 22 (2011) 1107-1110. |
| [17] | M.G. Ma, Y.J. Zhu, J. Chang, Monetite formed in mixed solvents of water and ethylene glycol and its transformation to hydroxyapatite, J. Phys. Chem. B 110 (2006) 14226-14230. |
| [18] | P. Zhang, Z.Y. Yang, S.X. Qiu, et al., Synthesis and characterization of poly (ethylene glycol)/hydroxyapatite hybrid nanomaterials, Chem. J. Chin. Univ. 33 (2012) 22-25. |
| [19] | G.A. Rodan, H.A. Fleisch, Bisphosphonates: mechanisms of action, J. Clin. Invest. 97 (1996) 2692-2696. |
| [20] | S. Boissier, M. Ferreras, O. Peyruchaud, et al., Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases, Cancer Res. 60 (2000) 2949-2954. |
| [21] | M. Naves, L. Gano, N. Pereira, et al., Synthesis, characterization and biodistribution of bisphosphonates Sm-153 complexes, correlation with molecular modeling interaction studies, Nucl. Med. Biol. 29 (2002) 329-338. |
| [22] | S. Boissier, S. Magnetto, L. Frappart, et al., Bisphosphonates inhibit prostate and breast carcinoma cell adhesion to unmineralized and mineralized bone extracellular matrices, Cancer Res. 57 (1997) 3890-3894. |
| [23] | P. Kαfarski, B. Lejczak, Aminophosphonic acids of potential medical importance, Curr. Med. Chem. Anti Cancer Agents 1 (2001) 301-312. |
| [24] | A. Zieba, G. Sethuraman, F. Perez, G.H. Nancollas, D. Cameron, Influence of organic phosphonates on hydroxyapatite crystal growth kinetics, Langmuir 12 (1996) 2853-2858. |
| [25] | D. Villemin, B. Moreau, A. Elbilali, et al., Green synthesis of poly(aminomethylenephosphonic) acids, Phosphorus Sulfur Silicon Relat. Elem. 185 (2010) 2511-2519. |
| [26] | L. Addadi, S. Raz, S. Weiner, Taking advantage of disorder: amorphous calcium carbonate and its roles in biomineralization, Adv. Mater. 15 (2003) 959-970. |
| [27] | X.Y. Zhou, Y.R. Jiang, C.C. Li, X.Y. Xie, Synthesis of poly(ethylene glycol)-functionalized hydroxyapatite organic colloid intended for nanocomposites, Chin. Chem. Lett. 24 (2013) 647-650. |
| [28] | B. Khorsand, G. Lapointe, C. Brett, J.K. Oh, Intracellular drug delivery nanocarriers of glutathione-responsive degradable block copolymers having pendant disulfide linkages, Biomacromolecules 14 (2013) 2103-2111. |
| [29] | Y.R. Cai, H.H. Pan, X.R. Xu, et al., Ultrasonic controlled morphology transformation of hollow calcium phosphate nanospheres: a smart and biocompatible drug release system, Adv. Mater. 19 (2007) 3081-3083. |
| [30] | K.W. Wang, Y.J. Zhu, X.Y. Chen, et al., Flower-like hierarchically nanostructured hydroxyapatite hollow spheres: facile preparation and application in anticancer drug cellular deliver, Chem. Asian J. 5 (2010) 2477-2482. |
| [31] | M. Uota, H. Arakawa, N. Kitamura, et al., Synthesis of high surface area hydroxyapatite nanoparticles by mixed surfactant-mediated approach, Langmuir 21 (2005) 4724-4728. |