




Mesoporous silica nanoparticles (MSNs) have attracted much attention for their potential use as drug delivery carriers because of their uniform pore size,large surface area,and high biocompatibility [1, 2, 3, 4, 5]. It has been demonstrated that both particle size and surface modification are critical parameters determining the blood half-life,opsonization,biokinetics,and biodistributions of MSNs [6, 7]. In general,large particle size (larger than 100 nm diameter) and poor particle stability (aggregation) result in rapid uptake by the reticuloendothelial system (RES) of the liver and spleen before the loaded drug reaches the cancer cells/tissues [8, 9]. The MSNs size should also be larger than 10 nm,as the tight endothelial junctions of normal blood vessels typically are of 5-10 nm in size [10]. Therefore,good control of the morphology,particle size, uniformity,and dispersity of MSNs is of considerable significance to their use in drug delivery.
On the other hand,surface properties are considered to be the most important aspect that influences the biocompatibility and blood half-life of MSNs. The surfaces of MSNs are often modified with poly(ethylene glycol) (PEG) polymers. Surface modification with PEG can form a hydrophilic layer around particles with increased dispersity and can greatly prevent protein adsorption on the nanoparticle surfaces [11]. PEG has been known to prevent protein adsorption on the nanoparticle surfaces,enhance circulation time,reduce nonspecific RES uptake,and facilitate preferential accumulation at tumor sites through the enhanced permeation and retention effect [11, 12, 13]. Although there have been several reports on the synthesis of PEGylated MSNs [14, 15, 16],it is still a challenge to synthesize PEGylated monodisperse MSNs with controllable sizes via a one-pot reaction. Furthermore,to facilitate clinical use and drug storage of MSNs,it is essential to develop a synthetic method to prepare redispersible PEGylated MSNs,which may be dried and resuspended later with no change in hydrodynamic size.
In this study,we report a one-pot synthesis of PEGylated MSNs with uniform shape,tunable sizes,and narrow size distributions. Particle synthesis was performed at high temperature (95℃) in aqueous solution in the presence of cetyltrimethylammonium bromide (CTAB) as the template,with tetraethylorthosilicate (TEOS) as the silica source,and triethanolamine (TEA) as the base catalyst. A polyethylene glycol silane (PEG-silane) was added directly into the synthesis batch to quench silica condensation on the particle surface. After removal of the template CTAB,these PEGylated MSNs exhibited excellent colloidal stability in biological media. More importantly,these PEGylated MSNs can be dried to a powdered solid and easily redispersed in biological media, maintaining their size for a period of at least 15 days. 2. Experimental
General: All chemicals were used without further purification. CTAB,TEOS,and TEA were purchased from Sigma-Aldrich. PEGsilane( MW 575-750 g/mol,8-12 EO,50% in ethanol) was obtained from Gelest (Morrisville,PA). Transmission electron microscopy (TEM) was carried out on a JEOL JEM-1400 instrument operated at 100 kV. Nitrogen adsorption-desorption isotherms were recorded on a Micromeritics ASAP 2020 Mautomated sorption analyser. The specific surface areas were calculated from the adsorption data in low pressure range using the Brunauer-Emmett-Teller (BET) model and pore size was determined following the Barrett- Joyner-Halenda (BJH) method. Fourier transform infrared (FTIR) spectra were collected by FTIR spectrometer (Perkin-Elmer 1760X) with the KBr method. Thermogravimetric analysis was carried out for powder samples using a TGA Q500 recorded from 100℃ to 800℃ in an air flow at a heating rate of 10℃/min. Dynamic light scattering (DLS) measurements were performed at 37℃ using a Malvern ZetaSizer NanoZS instrument,equipped with a He-Ne laser (633 nm) at a fixed scattering angle of 90°.
Synthesis of PEGylated MSNs (PEG-MSNs): CTAB (0.57 g) and TEA (80 mg) were added to distilled water (20 mL). The reaction mixture was heated to 95℃ under vigorous stirring. TEOS (1.5 mL) was then added dropwise to the solution. After 30 min,PEG-silane (1 mL) was added to the reaction mixture. After the solution was stirred at 95℃ for 1 h,it was cooled down to room temperature. The nanoparticles were collected by centrifugation,and washed several times with ethanol. In order to remove the template CTAB from the mesopores of the nanoparticles,they were suspended in a mixture solution of ethanol (12 mL) and concentrated HCl (1.5 mL, 36%),and the solution was treated three times by sonication in an ice bath for 30 min. Finally,PEG-MSNs were obtained by washing extensively with ethanol,and drying the nanoparticles from ethanolic suspensions using rotary evaporation. 3. Results and discussion
Common synthetic routes for the preparation of MSNs materials lead to particles with typical hydrodynamic diameters of 500 nm to several micrometers in water. In order to synthesize small MSNs,Bein et al. used the base TEA as a substitute for the more commonly used NaOH or NH4OH to synthesize MSNs with diameters of 20-150 nm [17]. TEA was thought to act as a complexing agent for silicate species and additionally as a growth inhibitor for mesoporous particles. However,the final product of this procedure is a gel of fused particles which cannot be redispersed in water after the drying process. In order to prepare small and redispersable MSNs,we further modified the Bein’s method by using the high reaction temperature (95℃) to accelerate the nucleation and the introduction of PEG-silane to terminate the silica condensation on the particle surface.
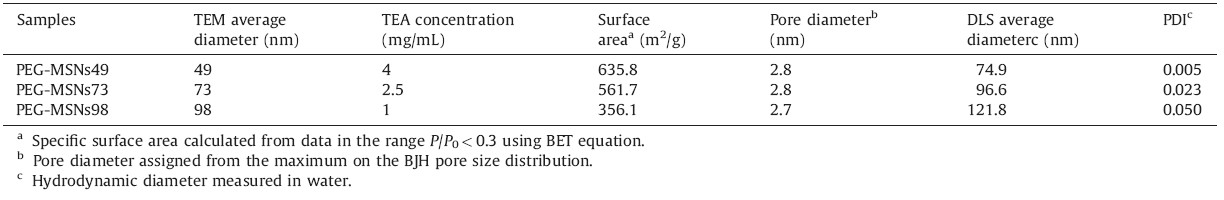
To investigate the effect of the PEG-silane addition on the MSNs synthesis,two samples were prepared as followed: unmodified MSNs and PEGylated MSNs. TEM images showed that both nanoparticles had similar diameters (~49 nm) and wormhole pore arrangement (Fig. 1A and B). For unmodified MSNs,silica connectors between nanoparticles were clearly observed. However, PEGylated MSNs are monodisperse nanoparticles with spherical shape. DLS measurements showed the hydrodynamic diameter of unmodified MSNs in water to be 100.1 nm (PDI 0.225) and PEGMSNs to be 74.9 nm (PDI 0.005) (Fig. 1C). The polydispersity value of PEG-MSNs (PDI 0.005) in water is close to zero,further confirming that the nanoparticles are monodisperse,uniform,and well-dispersed in water. These results indicated that the particle growth termination via the addition of PEG-silane quenched silica condensation on the particle surface,resulting in monodisperse nanoparticles.

|
Download:
|
| Fig. 1.TEM images of (A) unmodified MSNs,(B) PEG-MSNs,and (C) DLS measurements of unmodified MSNs and PEG-MSNs in water. | |
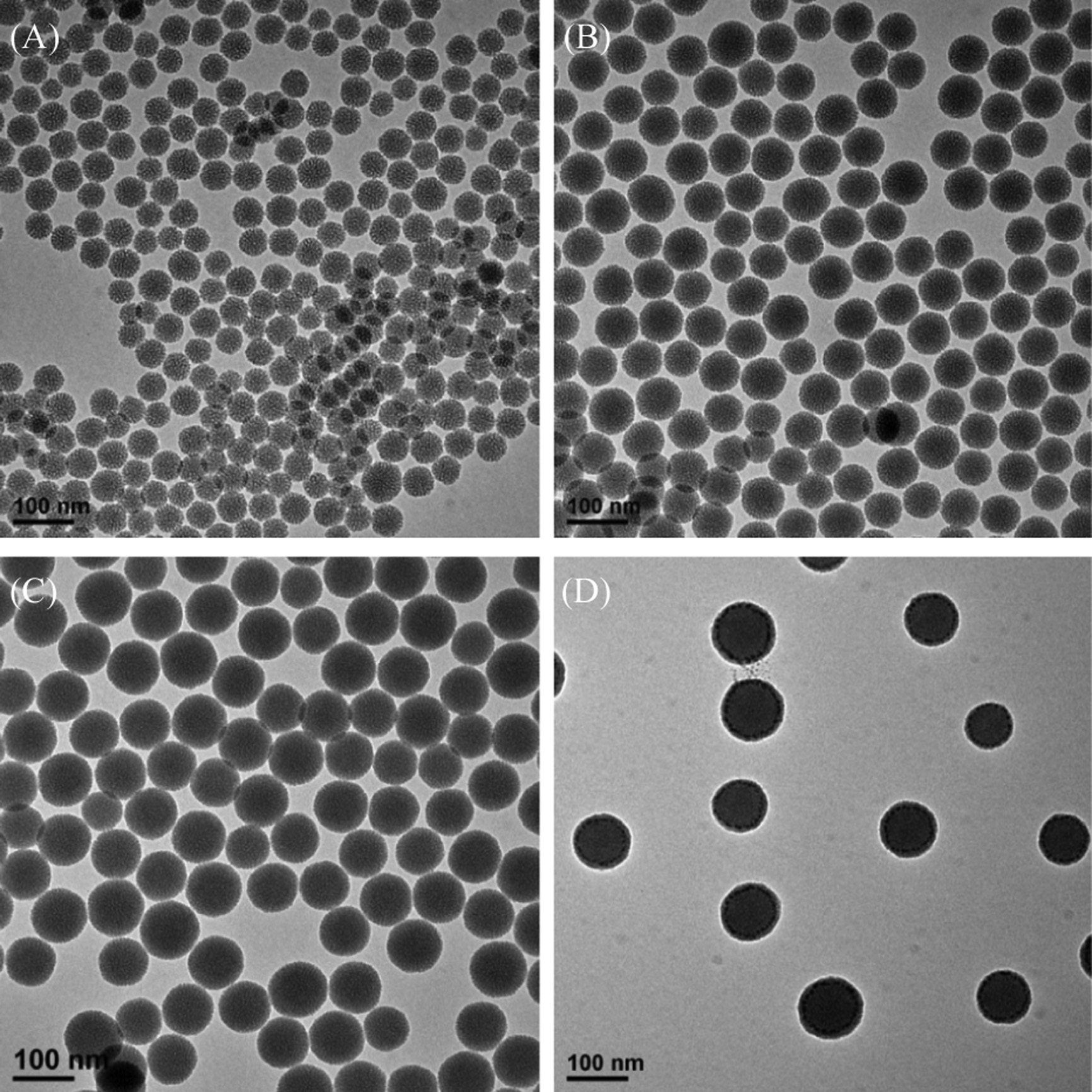
We systematically investigated the effects of the key reaction conditions (e.g.,amount of TEOS and PEG-silane,TEA concentration, reaction time) on the resulting PEG-MSNs,and found that the concentration of TEA is the dominant parameter influencing nanoparticle size. As the concentration of TEA decreased from 4 mg/mL to 1 mg/mL,the average diameter of nanoparticles increased from 49 nm to 98 nm (Fig. 2A-C). When the concentration of TEA decreased to as low as 0.75 mg/mL, nonporous silica nanoparticles were obtained (Fig. 2D). The nitrogen adsorption-desorption isotherm measurements showed that these PEG-MSNs with different diameters possessed relatively high specific surface areas of 356.1-635.8 m2/g and well-defined pore sizes of ~2.8 nm (Table 1 and Fig. S1 in Supporting information). Moreover,a very low surface area of 25.7 m2/g was observed for the nanoparticles formed at the TEA concentration of 0.75 mg/mL,further confirming their nonporous structure (Fig. S2 in Supporting information).

|
Download:
|
| Fig. 2.TEM images of PEG-MSNs that were obtained under the different concentration of TEA: (A) 4 mg/mL,(B) 2.5 mg/mL,(C) 1 mg/mL,and (D)0.75 mg/mL. | |
| Table 1 Physicochemical characteristics of PEG-MSNs with different diameters. |
{kind=link}
{kind=link}
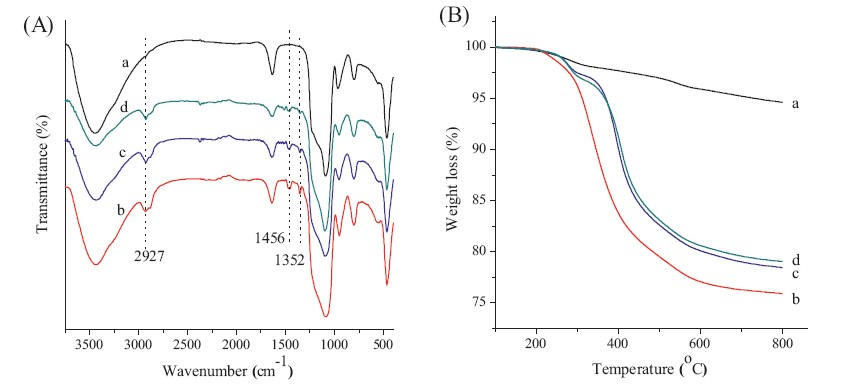
FTIR spectroscopy was used to confirm a successful template removal by complete disappearance of the C-H stretching vibrations between 2940 cm-1 and 2860 cm-1 and of the C-H bending vibration at 1488 cm-1 after the third extraction cycle (Fig. S3 in Supporting information). Furthermore,the successful surface modification of PEG-silane on the nanoparticles was confirmed using FTIR,as shown in Fig. 3A. For unmodified MSNs with 49 nm diameter (MSNs49),there was a very strong absorbance in the range of 1000-1200 cm-1,which can be attributed to Si-O-Si stretching in the mesoporous silica. Compared to MSNs49,several new bands were observed in the spectra of PEG-MSNs. The characteristic band at 2927 cm-1 was attributed to C-H stretching vibration of PEG-silane. The characteristic bands at 1456 cm-1 and 1352 cm-1 were assigned to CH2 and CH3 bending vibration of PEG-silane,respectively. Moreover, the ether band of PEG cannot be observed due to its overlap with the Si-O-Si stretching band in the spectra of PEG-MSNs.

|
Download:
|
| Fig. 3.(A) FTIR spectra and (B) TGA curves of (a) MSNs49,(b) PEG-MSNs49,(c) PEG-MSNs73,and (d) PEG-MSNs98. | |
{kind=link}
Thermogravimetric analysis also confirmed the successful PEGylated surface modification of MSNs. As shown in Fig. 3B, the weight loss of MSNs49 nanoparticles was about 5.3 wt% for the whole temperature range,which was largely due to the removal of absorbed chemical water and the dehydroxylation of silanol groups. For the PEG-MSNs49 nanoparticles,the weight loss was significant due to the PEG degradation at 280℃ [18]. The amount of PEG modified on the surface of MSNs was calculated on the basis of the TGA data,and they were found to be 24.1,21.5,20.9 wt% for PEG-MSNs49,PEG-MSNs73,and PEG-MSNs98,respectively.
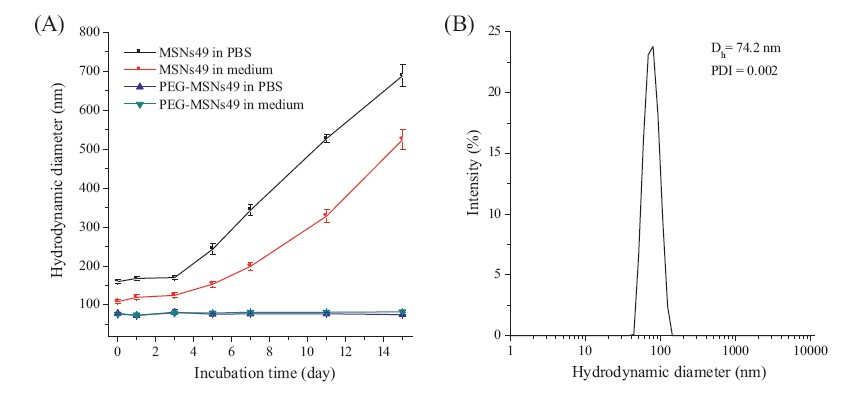
The dispersity and stability of PEG-MSNs were evaluated by DLS studies. As shown in Fig. 4A,PEG-MSNs49 can retain their narrow size distribution in biological media,including phosphate buffered saline (PBS,pH 7.4) solution and cell culture media (Dulbecco’s Modified Eagle’s Medium + 10% fetal bovine serum,DMEM + 10% FBS) at 37℃. However,the hydrodynamic size of MSNs49 in PBS or cell culture media increases over time and reaches the maximum values after 15 days. This result showed that MSNs49 formed irreversible aggregates in biological media. PEG-MSNs49 did not increase in size at all over a 15 day incubation period. In addition, PEG-MSNs49 can retain their narrow size distribution (PDI 0.002) even after drying the nanoparticles from ethanolic suspensions using rotary evaporation and simply redispersing the nanoparticle powder in cell culture media by ultrasonication for 5 min (Fig. 4B). All these results revealed that the PEGylated modification on MSNs significantly improved the nanoparticle dispersity and long-term stability in biological media.

|
Download:
|
| Fig. 4.(A) Long-term colloidal stability of MSNs49 and PEG-MSNs49 in the presence of PBS buffer solution (pH 7.4) or cell culture media containing 10% FBS at 37 8C. The error bars were obtained from three independent measurements. (B) Hydrodynamic diameter distribution of redispersed PEG-MSNs49 in cell culture media containing 10% FBS at 37 ℃. | |
{kind=link}
In summary,we have developed a one-pot method for the synthesis of PEGylated monodisperse MSNs with controllable particle size. In the synthesis of MSNs,the PEGylated step significantly reduces the aggregation and improves the colloidal dispersity of MSNs. These PEGylated MSNs exhibit excellent longterm stability in biological media,which might facilitate the use of these nanoparticles in drug delivery. Acknowledgment
This work was supported by the Self-determined Research Program of Jiangnan University (Nos. JUSRP11214 and JUSRP 51319B to JY). PAN
| [1] | B.G. Trewyn, S. Giri, I.I. Slowing, V.S. Lin, Mesoporous silica nanoparticle based controlled release, drug delivery, and biosensor systems, Chem. Commun. (31) (2007) 3236-3245. |
| [2] | Q.J. He, J.L. Shi, Mesoporous silica nanoparticle based nano drug delivery systems: synthesis, controlled drug release and delivery, pharmacokinetics and biocompatibility, J. Mater. Chem. 21 (2011) 5845-5855. |
| [3] | F.Q. Tang, L.L. Li, D. Chen, Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery, Adv. Mater. 24 (2012) 1504-1534. |
| [4] | Y.Y. Pu, Y. Li, W. Zhuang, et al., Preparation and characterizations of helical mesoporous silica nanorods using CTAB and alcohols, Chin. Chem. Lett. 23 (2012) 1201-1204. |
| [5] | Q. Zhang, F. Liu, K.T. Nguyen, et al., Multifunctional mesoporous silica nanoparticles for cancer-targeted and controlled drug delivery, Adv. Funct. Mater. 22 (2012) 5144-5156. |
| [6] | S.H. Wu, Y.S. Lin, Y. Hung, et al., Multifunctional mesoporous silica nanoparticles for intracellular labeling and animal magnetic resonance imaging studies, Chem- BioChem 9 (2008) 53-57. |
| [7] | Q.J. He, Z.W. Zhang, F. Gao, Y. Li, J.L. Shi, In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: effects of particle size and PEGylation, Small 7 (2011) 271-280. |
| [8] | K.M.L. Taylor, J.S. Kim, W.J. Rieter, et al., Mesoporous silica nanospheres as highly efficient MRI contrast agents, J. Am. Chem. Soc. 130 (2008) 2154-2155. |
| [9] | J. Kim, J.E. Lee, J. Lee, et al., Magnetic fluorescent delivery vehicle using uniform mesoporous silica spheres embedded with monodisperse magnetic and semiconductor nanocrystals, J. Am. Chem. Soc. 128 (2006) 688-689. |
| [10] | B. Haley, E. Frenkel, Nanoparticles for drug delivery in cancer treatment, Urol. Oncol.: Semin. Orig. Invest. 26 (2008) 57-64. |
| [11] | F. Danhier, O. Feron, V. Pré at, To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery, J. Controlled Release 148 (2010) 135-146. |
| [12] | Q.J. He, J.M. Zhang, J.L. Shi, et al., The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses, Biomaterials 31 (2010) 1085-1092. |
| [13] | H. Meng, M. Xue, T. Xia, et al., Use of size and a copolymer design feature to improve the biodistribution and the enhanced permeability and retention effect of doxorubicin-loaded mesoporous silica nanoparticles in a murine xenograft tumor model, ACS Nano 5 (2011) 4131-4144. |
| [14] | V. Cauda, C. Argyo, T. Bein, Impact of different PEGylation patterns on the longterm bio-stability of colloidal mesoporous silica nanoparticles, J. Mater. Chem. 20 (2010) 8693-8699. |
| [15] | V. Cauda, A. Schlossbauer, T. Bein, Bio-degradation study of colloidal mesoporous silica nanoparticles: effect of surface functionalization with organo-silanes and poly(ethylene glycol), Microporous Mesoporous Mater. 132 (2010) 60-71. |
| [16] | Y.S. Lin, N. Abadeer, C.L. Haynes, Stability of small mesoporous silica nanoparticles in biological media, Chem. Commun. 47 (2011) 532-534. |
| [17] | K. Möller, J. Kobler, T. Bein, Colloidal suspensions of nanometer-sized mesoporous silica, Adv. Funct. Mater. 17 (2007) 605-612. |
| [18] | Q. Zhang, C.H. Wang, L. Qiao, H.S. Yan, K.L. Liu, Superparamagnetic iron oxide nanoparticles coated with a folate-conjugated polymer, J. Mater. Chem. 19 (2009) 8393-8402. |